Ensembl: Collecting Orthologs |
Download the working directory, EnsOrthoCollection.tar.gz, including example files and Perl scripts.
| Download ortholog information from BioMart |
Enter the Ensembl top page and choose BioMart from the top line of this site.
Human protein gene IDs
Push "Dataset" from the blue left column and select Database. Now I chose "Ensembl Genes69" and "Homo sapiens genes (GRCh37.p8)".
Then push "Filters" from the left column and check "ID list limit" in the "GENE" column. Pull down the bar to "Ensembl Gene ID(s) [e.g. ENSG000xxx]" as follows. In the BioMart, you always need to check your setting in the left blue column (Dataset, Filters, Attributes, etc).
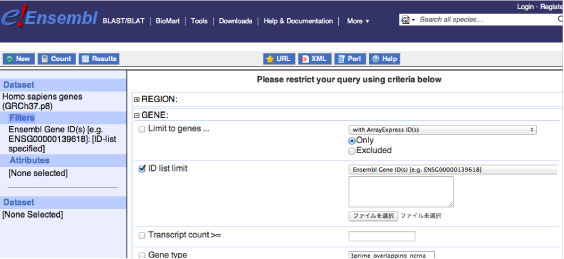
Push "Attribute" from the left column and select "GENE". Then select "Ensembl Gene ID" from "Ensembl" column as follows:
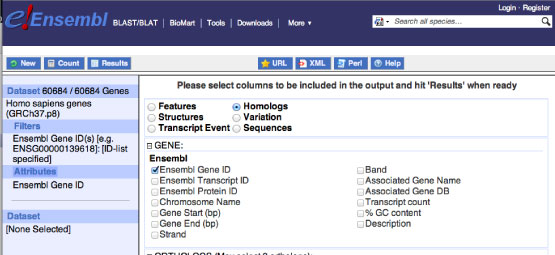
Then press "Results" button from the upper left column and download the mart_export.txt file including all Human protein coding gene IDs. Make sure you checked the two "Unique results only" buttons as follows.
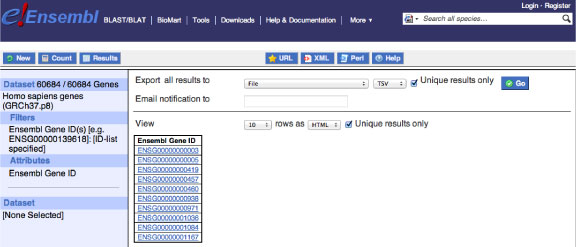
Orthologous protein gene IDs from the other species
Then we will collect the orthologous gene IDs of the species against human gene. Press (or Make sure to press) "Attributes" button from the left column and check "Homologs" in the upper column. For Chimp, chose "Chimp Ensembl GeneID" and "Homology Type" as follows. Then check the number of genes and download the mart_export.txt file as in the human section (see above). Check the downloaded file. The format should be like "EnsOrthoCollection/mart_export_ChimpTEST.txt".
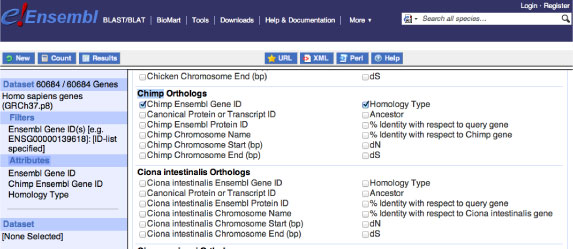
| Download databases |
For example, you can download the human cDNA database from
"homo_sapiens/cdna/Homo_sapiens.GRCh37.69.cdna.all.fa.gz"
and corresponding amino acid database from
"homo_sapiens/pep/Homo_sapiens.GRCh37.69.pep.all.fa.gz"
Download cDNA and amino acid databases for Pan troglodytes (Chimp) and Canis familiaris (dog) as shown above. Save the downloaded 6 files (AA/cDNA databased for 3 spp.) into the EnsOrthoCollection directory.
| Merge ortholog information file |
Merge ortholog information of multiple species
Go enter the downloaded file, EnsOrthoCollection, using terminal (Mac). Open 010_orthoTableMaker.pl file using your editor. Type the following command:
perl 010_orthoTableMaker.pl 010_out_orthoFile.txt mart_export_HumanTEST.txt
Make sure the above should be written in one line in your terminal. Then try 2nd and 3rd commands written in 010_orthoTableMaker.pl.
Sort multiple ortholog information
For example, if you want to choose only human genes including orthologs from all chosen species, you can read the output file from the above, "010_out_orthoFile_1_1_1.txt", by Excel and sort lines. Then save as the text file by tab delimited. In this case, changed the line break from Mac/Win to Unix using your editor. I saved as "020_orthoFile.txt" in my example.
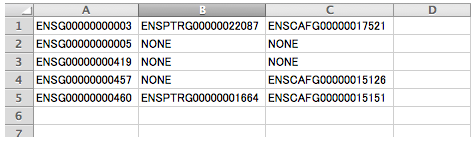
| Retrieve amino acid sequences |
We will retrieve the amino acid sequences from downloaded databases according to the orthology table (020_orthoFile.txt). Note that this program automatically chose longest transcript (amino acid sequences) among the recored with same gene ID. Before conduct the following analysis, change the name of the output file, 020_out_multiProtFiles, to something or just delete if it exists.
Type,
perl 020_longestPepPicker.pl 020_orthoFile.txt Homo_sapiens.GRCh37.69.pep.all.fa ENSG
020_orthoFile.txt: Orthology information file.
Homo_sapiens.GRCh37.69.pep.all.fa: Amino acid database.
ENSG: Alphabet including gene ID of each species. See the downloaded file.
Then try 2nd and 3rd commands shown in "020_longestPepPicker.pl".
| Retrieve cDNA sequences |
According to the amino acid file including several species, we will retrieve cDNA sequences from the database. Rename or delete 030_out_multiCDNAFiles if it exist.
Type,
perl 030_cDNApicker.pl 020_out_multiProtFiles Homo_sapiens.GRCh37.69.cdna.all.fa ENSG
020_out_multiProtFiles: Directory including amino acid files.
Homo_sapiens.GRCh37.69.cdna.all.fa: Downloaded cDNA database for Human.
ENSG: Alphabet including gene ID of each species.
| Align amino acid sequences |
Align all amino acid files using MAFFT. Install MAFFT before conduct the following command. You need to use MAFFT from the current directory. Otherwise rewrite the corresponding line of the following program.
Type
perl 040_autoMafft.pl 020_out_multiAAFiles
020_out_multiAAFiles: Directory including amino acid files.
| Align cDNA sequences |
Align cDNA sequences according to the aligned amino acid sequences using PAL2NAL. PAL2NAL is included in this example directory. PAL2NAL automatically assigns the corresponding codon sequence even if the input DNA sequence contains UTRs, polyA tails.
Type,
perl 050_autoPAL2NAL.pl 040_out_aligneAAfileDir 030_out_multiCDNAFiles
040_out_aligneAAfileDir: Directory including aligned amino acid files.
030_out_multiCDNAFiles: Directory including non-aligned cDNA files.
For, "2. ENSG00000000005.txt." and "3. ENSG00000000419.txt.", PAL2NAL will return the error message as follows:
ERROR: number of input seqs differ (aa: 0; nuc: 1)!!
It means no sequence was found in amino acid file. In this example, these 2 human genes do not have any orthologs and we got right answer about them.
| All in one analysis |
Type,
sh 000_allCommands.sh
I am not sure this works on Windows.