|
|
2015 年 1 月 20日 改訂
|
Newbler は NGS から得られたゲノムデータを解析するアセンブラー (Roche 社) です.このページの解説は,主に Linux machine (Scientific Linux) を Command Line Interface (CLI, Linux では端末エミュレータでコマンドを入力) で操作しています.私は Illumina 社 MiSeq のデータ解析に使っています.
Newbler の良い点は,あとでデータを追加して解析できる (incremental assembly, マニュアル PartC, p43,see below) 点だそうです.
Trinity との比較
Trinity はトランスクリプトームデータ解析に特化しているのに対し,Newbler はゲノムデータの解析もできます.また前者が K-mer assembly を行うのに対し,Newbler は overlap assembly を行うそうです.得られるリードが 150bp よりも長い場合は,Trinity より Newbler の方が良いそうです.NGS で出力されるリードは年を経るごとに長くなる傾向にあるので,Newbler を使うことが多くなるのではと思っています.
Trimming
ソフト自体に quality をチェックする機能がついているので,Trinity のようにデータの前後処理を行う必要がないようです.このため Trinity 解析のページで紹介した Solexa QA 2 (質の悪いデータを取り除く) や filterPCRdupl.pl (PCR duplicate を取り除く) などの解析は必要ないです.
以前,上のようなことを書きましたが,Trimming が必要という意見もありました.というのも,それぞれ 400 万リードからなるペアデータを Newbler で解析すると,数日たっても解析が終わらないことがあります (試しに 1.0*10^5 までリードを減らすと,数分で解析は終わりました).Trimmomatic などで Trimming した後に Newbler で解析すると良いそうです.
操作
以下ではコマンドラインで行う操作 (CLI: command line interface) を解説します.Version 29 です.マニュアルは P43 から解説がスタートします.
|
|
- まだ使って数週間ですが,リード数が多すぎるといつまで待っても解析が終わらない気がします.今のところ Ones step でも Increment Assembly でも症状は似ています.Solexa と Condetri からなる Trimming を行った後の 1.0*10^6 のペアリードは 3 時間で終わりましたが,Trimming なしの 5.3*10^6 ペアリードは一週間待っても解析が終わりません (2013 年 12 月).
- Input data の扱いについては,マニュアル P9 「Overview of the GD De Novo Assembler」に書かれています (2015 年 1 月).
|
|
Roche 社の HP にある「Request a free download of the software」から登録を行うことで,Free でダウンロードできます.
マニュアルはダウンロードして得られる以下の PDF ファイルです.ここでは De Novo Assembling をコマンドラインで行うので,P43 からを参照しています.オプションの説明は P228 からです.
USM-00058.09_454SeqSys_SWManual-v2.9_PartC.pdf
|
|
スパコン上に local でインストールするやり方を説明します.ここでは OIST の TOMBO というクラスターにインストールしていますが,他のスパコンでも状況は似ていると思います.ログインモードでインストールしてください.qlogin すると,インストールできませんでした.
解凍して得られた DataAnalysis_2.9_All に入り
sh setup.sh
と入力してください.
|
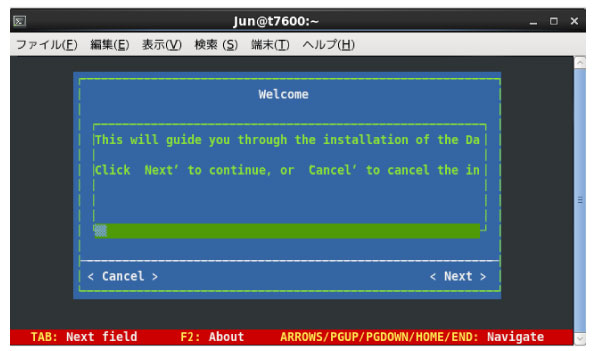 |
上のような画面が表示されます.tab でカーソル (長い横棒) を移動して,Enter で Next を選択します.
|
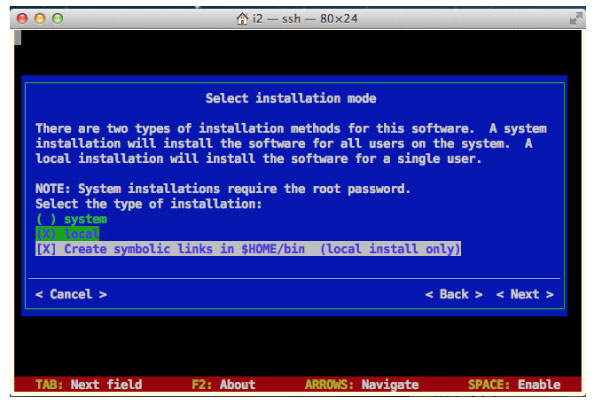 |
system ではインストールできませんでした.↓キーで local を選んでインストールしました.
|
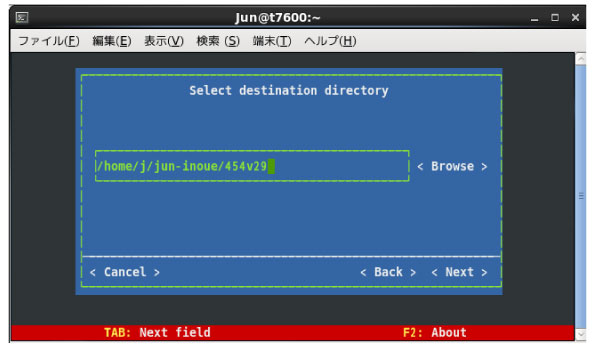 |
インストールするディレクトリは,こちらでつけた名前をずっと使います.私が操作した範囲では,インスールした後にディレクトリの名前を変えたり場所を bin に入れたりすると,たとえ PATH を正確に切ったりコマンドでディレクトリのアドレスを入力しても,解析の際に使用するプログラムを見つけてくれませんでした.
上であるように,454 を 454v29 に変更しても解析はうまく動くことは確認しました.
あとは画面の指示に従ってください.
|
エラー対策
購入したばかりの Linux machine (Scientific Linux) に Newbler をインストールしようとしたら,以下のようなメッセージが出ました.
「One or more of the following libraries was not found
zlib.i686 libXi.i686 libXtst.i686 libXaw.i686 」
これを無視してインストール手順を完了したのですが,デスクトップに作成されたアイコンをダブルクリックしてもプログラムが起動しませんでした.そこで,Linux に詳しい友人にインストール手順の解決を頼みました.以下の手順で必要なもののインストールを行ったようです.
「ia32-libs fedora」で Google 検索.以下のページを参照していました.
http://seqanswers.com/forums/showthread.php?t=8944&page=2
https://ask.fedoraproject.org/question/9556/how-do-i-install-32bit-libraries-on-a-64-bit-fedora/
http://stackoverflow.com/questions/8328250/centos-64-bit-bad-elf-interpreter
http://www.pressingquestion.com/10933426/Trying-To-Install-I386-Library
とりあえず Wine をインストール.こちらによると Wine とは Linux 上で Windows を動作させるプログラム群のようです.
yum install wine
Wine によって Windows が走るか確認.
winecfg
ライブラリをインストール.ファイル名の間はスペースです.
yum install zlib.i686 libXi.i686 libXtst.i686 libXaw.i686
|
|
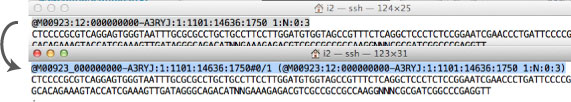
newbler で用いられている fastaq 形式の header は,最新機種から得られる形式に対応していません.このため,old-style.fastq に書き換えが必要な場合があります.こちらを参照しました.以下のコマンドを実行すれば良いです.その際,new-style.fastq (MiSeq などから得られたファイル) と old-style.fastq (newbler 解析用の,新たに得られるファイル) を適切な名称に変更します.
cat new-style_.fastq | awk '{if (NR % 4 == 1) {split($1, arr, ":"); printf "%s_%s:%s:%s:%s:%s#0/%s (%s)\n", arr[1], arr[3], arr[4], arr[5], arr[6], arr[7], substr($2, 1, 1), $0} else if (NR % 4 == 3){print "+"} else {print $0} }' > old-style.fastq
注意: header に問題がある場合,Newbler は配列の pair を読み取らないため,Scaforld を作成しないそうです.ただし,-cdna オプションを用いた場合は,scafold はこれに関わらず作成されないようです (マニュアル P58).
|
|
|
トランスクリプトームデータの解析を行います.
testNB_cDNA_runAssembly.tar.gz
例題で行っているコマンドを説明します.「sh jobCommand.sh」で実行します.ここでは上述の「fastaq file の書き換え」の説明と操作は省略しています.
runAssembly -cdna -cpu 12 -het -siod -qo -m musR1e4.fastq musR2e4.fastq
-cpu 12: cpu の数を設定します (マニュアル P231).
-cdna: cDNA/ Transcriptome assembly を行います (マニュアル P53).
-m: マニュアル P217 参照.
musR1e4.fastq musR2e4.fastq: マニュアル P217 参照.インファイルです.Paired end と指定しなくても,勝手にペアを判断してくれるようです.なお,インファイルは複数のファイルを指定可能なようです.
注意: Incremental Assembly (下記) と異なり,アウトファイルの名前は指定できないようです.
スクリーンアウトは以下のようになるはずです.エラーが出ていないか気をつけてください.メモリーが足りない,というメッセージが出ることがあります.
さらに,こちらにあるように,配列が paired で読み取れているかどうか,確認してください.以下に示すように,paired で読み込まれた場合は,太字の marker as matepairs が表示されるはずです.paired で読まれない場合は,インファイルの head が正しいかどうか,よく確認してください.
アウトファイルについては下の方のコラムに書きました.
[Jun@t7600 testNB_cDNA_runAssembly]$ sh jobCommand.sh
Created assembly project directory P_2013_11_25_10_08_11_runAssembly
2 read files successfully added.
musR1e4.fastq (Illumina paired-end dataset, with standard scores)
musR2e4.fastq (Illumina paired-end dataset, with standard scores)
Assembly computation starting at: Mon Nov 25 10:08:11 2013 (v2.9 (20130529_1641))
Indexing/Screening musR2e4.fastq (with quality scores)...
-> 9886 reads, 1966282 bases, 9670 marked as matepairs.
Indexing/Screening musR1e4.fastq (with quality scores)...
-> 9963 reads, 1956016 bases, 9854 marked as matepairs.
Uploading reads into memory...
-> 19524 of 19524
Setting up long overlap detection...
-> 19524 of 19524, 11880 reads to align
Building a tree for 99412 seeds...
Computing long overlap alignments...
-> 19524 of 19524
Setting up overlap detection...
-> 19524 of 19524, 15770 reads to align
Starting seed building...
-> 19524 of 19524
Building a tree for 218278 seeds...
Computing alignments...
-> 19524 of 19524
Checkpointing...
Detangling alignments...
-> Level 3, Phase 9, Round 2...
Checkpointing...
Building contigs/isotigs...
-> 68 large contigs, 330 all contigs
-> 314 isogroups, 323 isotigs
Computing signals...
Checkpointing...
Generating output...
-> 148842 of 148842...
Assembly computation succeeded at: Tue Jan 20 19:08:30 2015
[jun-inoue:testNB_runAssembly]$
-cpu オプション
-cpu 0 だとすべての CPU を自動的に使ってくれるそうです (マニュアル p231).以下実験してみたところ,確かに早くなりました.「システム・モニタ」(下図) で見ても,すべての CPU が使われていますし,音もうるさくなりました.テスト Examples はこちらです cpuTest.tar.gz (29MB).-batchsize オプションを使うやり方もあるようですが,今のところよくわからないです.
#runAssembly -cpu 0 -batchsize 1 -cdna musR1_e5.fastq musR2_e5.fastq
#=> 1min 53sec
#runAssembly -cpu 0 -cdna musR1_e5.fastq musR2_e5.fastq
#=>1min 50 sec - 1min 59sec
#runAssembly -cpu 6 -cdna musR1_e5.fastq musR2_e5.fastq
#=>2min 7sec
|
|
| 例題2: Incremental Assembly |
|
ここでは Incrementaly assembly という方法でトランスクリプトームデータの解析を行います.マニュアルによるとこの方法は,すでに存在する assembly に read を加えてゆく方法です.
実は私はまだ Incrementaly assembly をやったことがありません.とくに Incrementary でなくても解析できるようなので,まずは incrementary でない通常の解析をやったので紹介します.
testNB_cDNA.tar.gz
jobTest.sh に入っていて,適宜文頭の「#」をはずして「sh jobTest.sh」で実行します.「fastaq file の書き換え」の説明と操作は省略しています. 解析を行う前に,「testAssembl」というディレクトリの名前を変更するか削除してください.
プロジェクトの開始を宣言
newAssembly -cdna testAssembl
-cdna: cDNA/ Transcriptome assembly を行います (マニュアル P53).
testAssembl というディレクトリが作成されます.すでにあると解析が走りません.
既存のプロジェクトに配列を加える
addRun -p -lib shortPE testAssembl *.fastq
-p -lib: paired シーケンスを認識させるようです (マニュアル P240).
Assembly を行い結果を出力する
runProject -cpu 12 -het -siod -qo -m testAssembl
-large: ゲノムアセンブリの時に使います.トランスクリプトームデータの解析でつけると,無理に Contig をつなげてしまうようです.
スクリーンアウトは以下のようになるはずです.
[jun-inoue:testNB_cDNA]$ sh jobTest.sh
Created assembly project directory testAssembl
2 read files successfully added as explicit paired-end files.
mus1.fastq (Illumina paired-end dataset, with standard scores)
mus2.fastq (Illumina paired-end dataset, with standard scores)
Assembly computation starting at: Wed Nov 20 10:53:10 2013 (v2.9 (20130529_1641))
Indexing/Screening mus2.fastq (with quality scores)...
-> 9972 reads, 2261631 bases, 9492 marked as matepairs.
Indexing/Screening mus1.fastq (with quality scores)...
-> 9960 reads, 2216884 bases, 9811 marked as matepairs.
Uploading reads into memory...
....
(省略)
....
Generating output...
-> 149834 of 149834...
Assembly computation succeeded at: Wed Nov 20 10:53:20 2013
|
|
マニュアル P 55 以降にまとめられています.One-step assembly で解析を行った場合,結果は P_yyyy_mm_dd_hh_min_sec_runAssembly/assembly というディレクトリに保存されます.
454AllContigs.fna
作成されたコンティグが保存されています.
454Scaffolds.fna
スキャフォールドです.-cdna オプションを選んでいると作成されません (マニュアル p58).
454NewblerProgress.txt
ログが記録されます.これを見れば,解析がどこまで進んだかわかります.内容は cat で見た方が良いです.vi や less で見ると,表示がおかしいです (改行コードの問題?).
|
|
Introduction to Newbler - The GenePool
パワーポイントスライドです.
Blog for newbler
Newbler に関するブログです.Newbler の概要を知らないで読んでも,あまり意味が分からないかもしれません.ブログなので,「Read the rest of this entry」(注意: プリントアウトされない) を押さないと全文が読めないです.
University of Arizona
Platanus
IDBA
メタゲノム解析に用いるらしい.ハプロイド専用?
|
|
このページは主に OIST に所属する研究者の方から教えていただいた情報をもとに作成しています.皆さんのご協力に感謝します.
|
|