listup_refseq_ncbi_cli.sh.zip
STEP 0 —(任意)conda 環境を有効化
conda activate ncbi_datasets
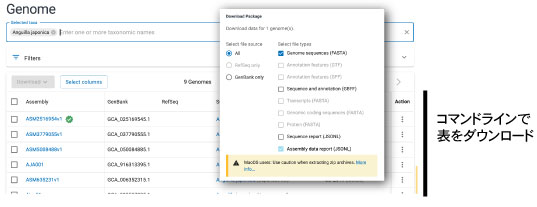
STEP 1 — メタデータだけを軽量で取得する
(※ 実データを除外するために --include none を指定)
datasets download genome taxon "<TAXON_NAME>" --include none --filename <TAXON>.zip
例:
datasets download genome taxon "Cyprinoidei" --include none --filename cyprinoidei.zip
--include none により、データ本体(fasta, gff など)が除外され、
metadata.jsonl だけの軽量 ZIP(数百 KB〜数 MB) が得られます。
(NCBI Datasets が JSON Lines のメタデータを含む ZIP を作る仕様
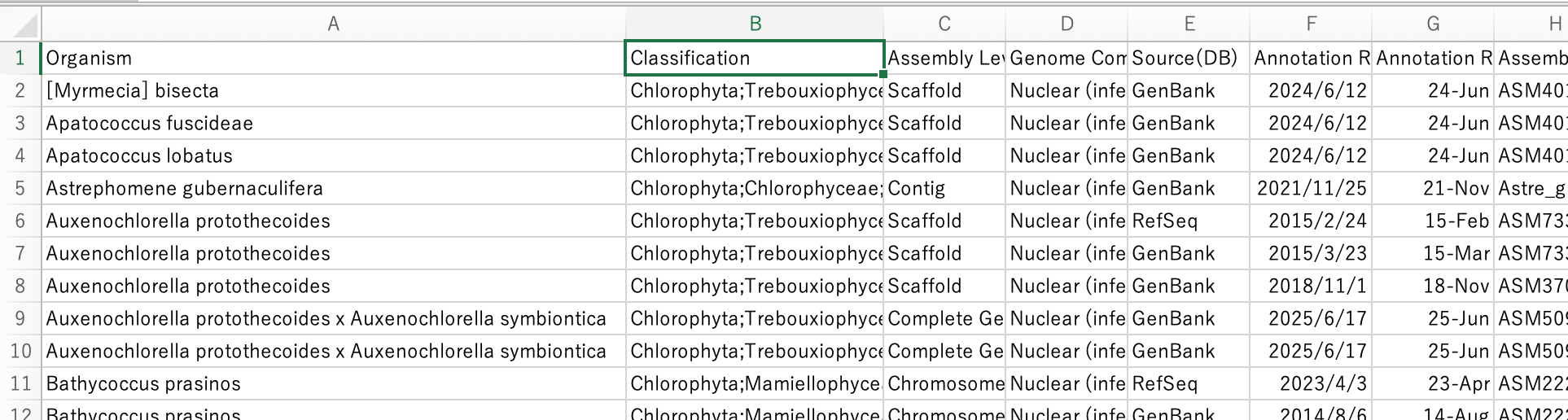
STEP 2 — 最低限の情報を TSV に変換する
(organism-name / assembly name / accession)
dataformat tsv genome \
--package <TAXON>.zip \
--fields organism-name,assminfo-name,accession \
> <TAXON>.tsv
例:
dataformat tsv genome --package cyprinoidei.zip \
--fields organism-name,assminfo-name,accession \
> cyprinoidei.tsv
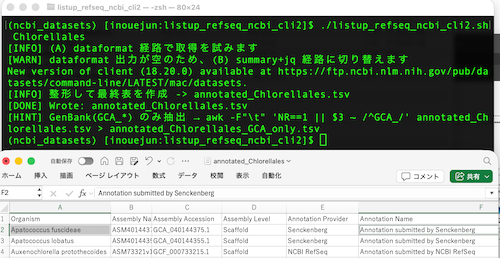
STEP 3 — RefSeq(GCF_)だけ抽出する
RefSeq は NCBI の規則で アクセッションが GCF_ ではじまるため、最も確実なフィルタ方法です(GenBank は GCA_)。
これは NCBI Datasets metadata でも公式に提供されるフィールド。
awk -F '\t' 'NR==1 || $3 ~ /^GCF_/' <TAXON>.tsv > <TAXON>_refseq.tsv
例:
awk -F '\t' 'NR==1 || $3 ~ /^GCF_/' cyprinoidei.tsv > cyprinoidei_refseq.tsv
(任意)STEP 4 — 詳細フィールドが必要なら catalog で確認
ZIP 内の metadata.jsonl に含まれるすべてのフィールドは:
dataformat catalog --package <TAXON>.zip
で確認できます。
(Datasets v18 以降、この catalog は JSON-lines の構造一覧を返すようになっています。)
必要なフィールドがわかれば:
dataformat tsv genome --package <TAXON>.zip --fields <FIELD1>,<FIELD2>,<FIELD3>,... > out.tsv
の形で自由に選んで追加できます。
STEP 5 — (任意)Excel 形式で出力
dataformat excel genome \
--package <TAXON>.zip \
--fields organism-name,assminfo-name,accession \
> <TAXON>.xlsx
(Excel でそのまま並べ替え・フィルタできます)
|