|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
Œn“‰ًگح‚ةژg‚¦‚»‚¤‚ب Perl script ‚ً‚آ‚‚ء‚ؤ‚¢‚ـ‚·پDٹب’P‚بژQچlڈ‘‚₱‚؟‚ç‚جƒTƒCƒg‚ًژQڈئ‚µ‚ب‚ھ‚çژg‚ء‚ؤ‚‚¾‚³‚¢پD |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
’تڈي Mac ‚ة‚ح perl ‚ھƒCƒ“ƒXƒgپ[ƒ‹‚³‚ê‚ؤ‚¢‚ـ‚·پDˆب‰؛‚جƒRƒ}ƒ“ƒh‚ة‚و‚ء‚ؤپCƒoپ[ƒWƒ‡ƒ“‚ًٹm”F‚µ‚ؤ‚‚¾‚³‚¢.
ژ„‚حƒAƒbƒvƒOƒŒپ[ƒh‚µ‚½‚©‚ء‚½‚ج‚إ‚·‚ھپC‚â‚è•û‚ھ‚ي‚©‚炸 (ƒAƒbƒvƒOƒŒپ[ƒh‚ح–³‚¢‚ج‚©‚à‚µ‚ê‚ـ‚¹‚ٌ)پCŒ‹‹اƒCƒ“ƒXƒgپ[ƒ‹‚µ‚ـ‚µ‚½پD‚±‚؟‚ç‚©‚çچإگV”إ‚ًƒ_ƒEƒ“ƒچپ[ƒh‚µ‚ؤ‚‚¾‚³‚¢ (The latest releases in each branch ‚ئ‚¢‚¤•\‚إ‚·)پD‚½‚¾‚µپC5.13 ‚ب‚ا‚ج‚و‚¤‚ةٹïگ”‚حˆہ’肵‚ؤ‚¢‚ب‚¢‚و‚¤‚ب‚ج‚إپC‹ôگ”‚ً‘I‚ٌ‚¾•û‚ھ–³“ï‚إ‚·پDژ„‚ح 5.12.2 ‚ًƒ_ƒEƒ“ƒچپ[ƒh‚µ‚ـ‚µ‚½پD‰ً“€‚µ‚ؤ“¾‚ç‚ꂽƒtƒHƒ‹ƒ_‚ة“ü‚èپCˆب‰؛‚جڈ‡”ش‚إƒRƒ}ƒ“ƒh‚ً‚¤‚؟‚ـ‚·پD‘S‘ج‚إ 10 •ھˆت‚©‚©‚ء‚½‚إ‚µ‚ه‚¤‚©پD make test ‚à‚â‚é‚و‚¤‚ةژwژ¦‚³‚ê‚ـ‚·‚ھپC‚ا‚؟‚ç‚إ‚à—ا‚¢‚و‚¤‚إ‚·پDmake test ‚¾‚¯‚إ‚à 5 •ھˆت‚©‚©‚ء‚½‚و‚¤‚ب‹C‚ھ‚µ‚ـ‚·پD |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
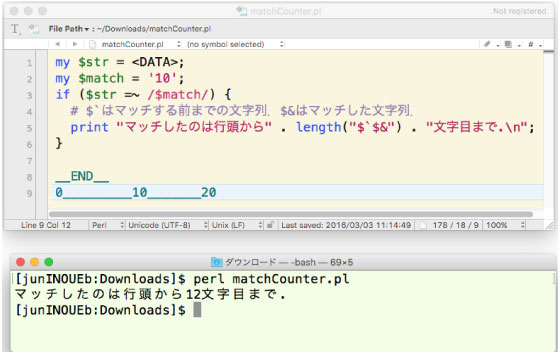
ˆب‰؛‚ً test.pl ‚ئ‚µ‚ؤ•غ‘¶‚µ‚ـ‚·پDƒ^پ[ƒ~ƒiƒ‹‚©‚çپCپuperl test.plپv‚ئ“ü—ح‚µ‚ؤ‚‚¾‚³‚¢پD
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
‰–ٹî”z—ٌˆبٹO‚ج•¶ژڑ‚ًژو‚èڈœ‚
ƒMƒƒƒbƒv‚ًژو‚èڈœ‚
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
•،گ”ƒtƒ@ƒCƒ‹“à•”‚ة‚ ‚é“ء’è‚ج•¶ژڑ‚ً•دچX‚·‚é‚ة‚حپC‚±‚؟‚ç‚جƒyپ[ƒW‚ًژQڈئ‚µ‚ؤ‰؛‚³‚¢پD
—ل 1) html ƒtƒ@ƒCƒ‹‚ج“à•”‚ً•دچX‚·‚éپD
ٹg’£ژq‚ھ .html ‚ة‚ب‚ء‚ؤ‚¢‚éƒtƒ@ƒCƒ‹“à•”‚ًŒںچُ‚µپC#ffffdd ‚ً #aaaacc ‚ة•دٹ·‚µ‚ـ‚·پD's/\n/\r/ig;' ‚ة‚·‚ê‚خپC‰üچsƒRپ[ƒh‚ً Unix ‚©‚ç Mac ‚ة•دٹ·‚µ‚ـ‚·پD |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
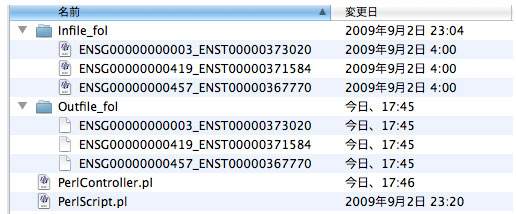
system ‚¨‚و‚ر opendir ٹضگ”‚ًژg‚ء‚ؤپC‘¼‚ج perl script ‚ً“®‚©‚µ‚ـ‚·.
PerlController.pl: ‘خڈغ‚ئ‚ب‚é perl scriptپD‘€چى‚·‚é script ‚ح INFILE ‚ئ OUTFILE ‚ً‰و–ت‚ة“ü—ح‚·‚éƒXƒNƒٹƒvƒg‚إ‚ ‚é•K—v‚ھ‚ ‚è‚ـ‚· (—ل: perl PerlScript.pl infile.fas > outfile)پD PerlScript.pl: p distance ‚ًŒvژZ‚·‚éƒvƒچƒOƒ‰ƒ€‚إ‚·پDPAUP ‚â PAML ‚ب‚ا‘¼‚جƒvƒچƒOƒ‰ƒ€‚ئ”نٹr‚µ‚ؤپC’چˆس‚µ‚ؤژg‚ء‚ؤ‚‚¾‚³‚¢پD PerlContoroller.pl |
||||||||||||||||||||||||||||||
 |
||||||||||||||||||||||||||||||
ژه‚ةˆب‰؛‚جƒeƒNƒjƒbƒN‚ًژg‚ء‚ؤ‚¢‚ـ‚·پD ƒAƒEƒgƒvƒbƒgƒfƒBƒŒƒNƒgƒٹ‚ًچىگ¬‚·‚éپD |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
“ٌ‚آ‚جƒtƒHƒ‹ƒ_‚ً”نٹr‚µ‚ؤپCڈ¬‚³‚¢ƒtƒHƒ‹ƒ_‚إ‘«‚è‚ب‚¢ƒtƒ@ƒCƒ‹‚ًƒٹƒXƒgƒAƒbƒv‚µ‚ـ‚·پD
MissigFile_fol.tar.gz |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
#!/usr/bin/perl |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| Unix ‚©‚ç Mac Œ`ژ®‚ة‰üچsƒRپ[ƒh‚ً•دٹ·‚µ‚ـ‚·پD LineBreakChange.tar.gz ژQچl‚ة‚ب‚éƒTƒCƒgپD http://osksn2.hep.sci.osaka-u.ac.jp/~taku/osx/crlf.html |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
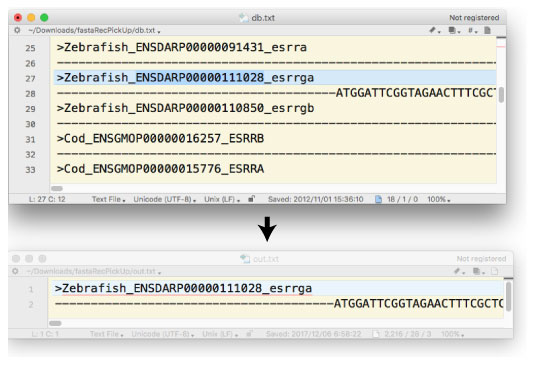
—ل‘è 1: ƒAƒŒƒC“à•”‚ةŒںچُڈًŒڈ‚ھ‚ ‚ê‚خ hit ‚ًڈo—ح‚·‚éپD
http://cast-a-spell.at.webry.info/200708/article_2.html
http://www.tohoho-web.com/wwwperl2.htm
ژہچsŒ‹‰ت‚حˆب‰؛‚ج’ت‚è.
* ƒپƒ^•¶ژڑ‚ً•¶ژڑ‚»‚ج‚à‚ج‚ئ‚µ‚ؤ”»’f‚³‚¹‚é‚ة‚ح m|\Qپ`\E| ‚ًژg‚¤پD
•â‘«. ƒپƒ^•¶ژڑ‚ً•¶ژڑ‚»‚ج‚à‚ج‚ئ‚µ‚ؤ”»’f‚³‚¹‚éژè–@‚حپC‚©‚ب‚è•ض—کپD |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| matchCounter.pl.tar.gz (2016 ”N 3 Œژ) ‚±‚؟‚ç‚ًژQڈئ‚µ‚ـ‚µ‚½پD |
||||||||||||||||||||||||||||||
 |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| fastaReadHash.tar.gzپ@(2017 ”N 12 Œژ) |
||||||||||||||||||||||||||||||
#!/usr/bin/perl -w # perl fasHash.pl db.fas > out.fas use strict; use warnings; my $infile = "db.fas"; open(INFILE,"$infile") or die "$!"; my @inFileCont = |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
 |
||||||||||||||||||||||||||||||
| fastaRecPickUpNoMemory.tar.gz (2017 ”N 12 Œژ) |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| fasToPhy2D.tar.gz | ||||||||||||||||||||||||||||||
#!/usr/bin/perl -w |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
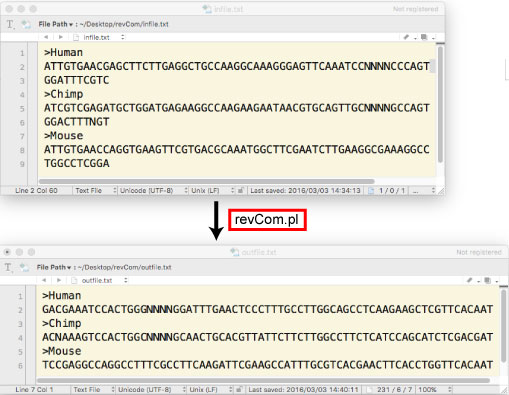
| revCom.tar.gz | ||||||||||||||||||||||||||||||
 |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
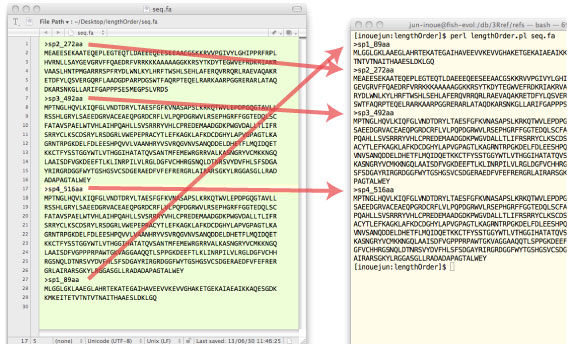
| lengthOrder.tar.gz (2013 ”N 6 Œژ) | ||||||||||||||||||||||||||||||
 |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
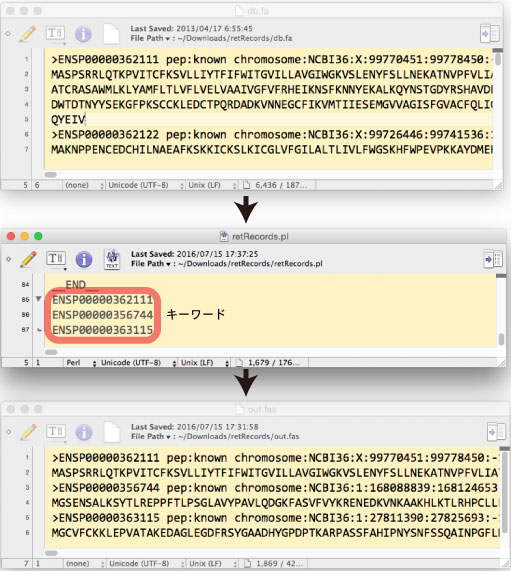
ƒLپ[ƒڈپ[ƒh‚ھ name line ‚ةٹـ‚ـ‚ê‚郌ƒRپ[ƒh‚ًƒfپ[ƒ^ƒxپ[ƒX‚©‚ç’ٹڈo‚µ‚ـ‚·پD |
||||||||||||||||||||||||||||||
 |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
fasta Œ`ژ®‚جƒtƒ@ƒCƒ‹‚©‚ç”z—ٌ‚ًگط‚èڈo‚µ‚ـ‚· (2017 ”N 7 Œژ)پD
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
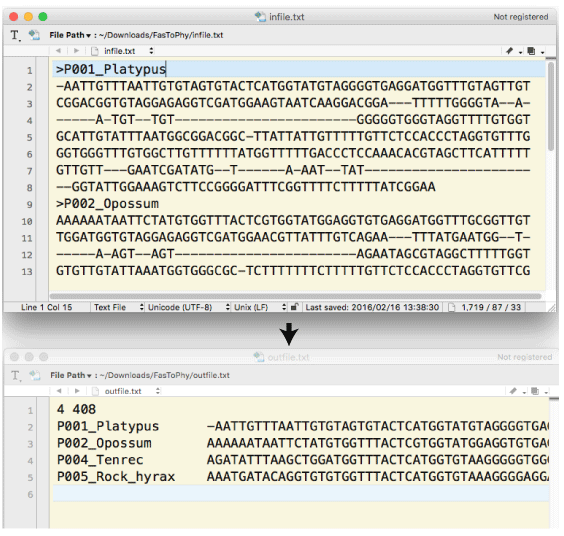
infile “à•”‚ة‚ ‚é OTU –¼‚ج•دٹ·‚ًچs‚¢پCoutfile ‚ةڈ‘‚«ڈo‚µ‚ـ‚·پD list ‚ة•شٹز‘O‚ج–¼‘O‚ئ•شٹزŒم‚ج–¼‘O‚ًƒٹƒXƒgƒAƒbƒv‚µ‚ؤ‚¨‚«‚ـ‚·پD FasNameChange_fol.tar.gz •،گ”‚جƒtƒ@ƒCƒ‹“à‚ج•¶ژڑ—ٌ‚ً•دچX‚·‚éڈêچ‡‚حپC‚±‚؟‚ç‚ھژQچl‚ة‚ب‚è‚ـ‚·پD |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
ƒٹƒXƒgƒAƒbƒv‚µ‚½OTU –¼‚ةڈ]‚ء‚ؤ sequence file ‚ً•ہ‚ر‘ض‚¦‚ـ‚·پDfasta file ‚ةƒٹƒXƒgƒAƒbƒv‚µ‚½ OTU –¼‚ھ‚ب‚¢ڈêچ‡‚حپC’Z‚¢ƒMƒƒƒbƒv”z—ٌ‚ھڈ‘‚©‚ê‚ـ‚·پDƒٹƒXƒgƒAƒbƒv‚µ‚½ OTU –¼‘O‚ئ sequence file ‚ج OTU –¼‚حˆê’v‚³‚¹‚ؤ‚‚¾‚³‚¢ [2010 ”N 12 Œژ]پD |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
—ل‚¦‚خپCˆâ“`ژq‚²‚ئ‚ةٹـ‚ـ‚ê‚é OTU گ”‚ھˆظ‚ب‚é‚ئ‚«‚ةپCٹـ‚ـ‚ê‚ؤ‚¢‚ب‚¢ OTU ‚ةƒMƒƒƒbƒv‚¾‚¯‚©‚ç‚ب‚é”z—ٌ‚ً‰ء‚¦‚ـ‚·پDƒnƒbƒVƒ…‚ًژg‚ء‚ؤ‚¢‚ـ‚·پDGapOTUAdder_fol.tar.gz
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
‘S‚ؤ‚جژي–¼‚ًƒٹƒXƒgƒAƒbƒv‚µ‚½ƒtƒ@ƒCƒ‹‚ًچىگ¬‚µ‚ؤ‚‚¾‚³‚¢پD‚±‚جƒٹƒXƒg‚ئƒtƒ@ƒXƒ^ƒtƒ@ƒCƒ‹‚جژي–¼‚حٹ®‘S‚ةˆê’v‚µ‚ؤ‚¢‚é•K—v‚ھ‚ ‚è‚ـ‚·پD”z—ٌ‚ھ–³‚¢ˆâ“`ژq‚حپC‘¼‚جژي‚ئ“¯‚¶’·‚³‚جƒMƒƒƒbƒv‚ھ‰ء‚¦‚ç‚ê‚ـ‚·پD‘ه‹K–حƒfپ[ƒ^‚ض‚ج“K—p‚ًچl‚¦‚ؤپCƒپƒ‚ƒٹپ[‚ًژg‚ي‚ب‚¢‚و‚¤‚ةƒCƒ“ƒtƒ@ƒCƒ‹‚©‚ç 1 ژي‚أ‚آ”z—ٌ‚ًژو‚èڈo‚µ‚ؤƒAƒEƒgƒtƒ@ƒCƒ‹‚ةڈ‘‚«ڈo‚µ‚ـ‚·پD”z—ٌ‚حƒAƒ‰ƒCƒپƒ“ƒg‚³‚ê‚ؤ‚¢‚é•K—v‚ھ‚ ‚è‚ـ‚· [2010 ”N 12 Œژ]پD
SeqConcatBD_fol.tar.gz |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| fasToPhy.tar.gz (2016 ”N 3 Œژ)پD | ||||||||||||||||||||||||||||||
 |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
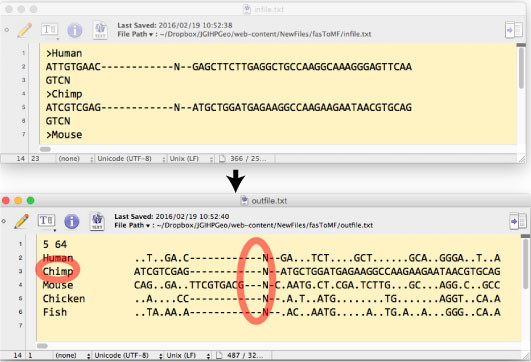
| fasToMF.tar.gz [2016 ”N 3 Œژ] | ||||||||||||||||||||||||||||||
 |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| fasToMF_return.tar.gz [2016 ”N ‚V Œژ] | ||||||||||||||||||||||||||||||
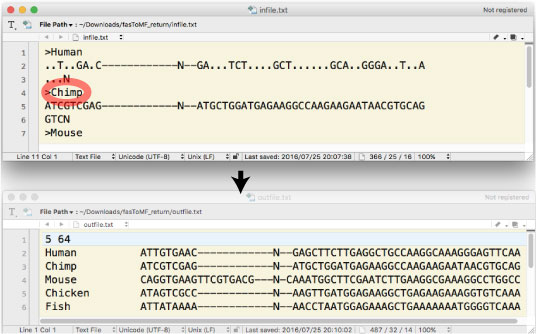 |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| fasToPhy_num.tar.gz (2016 ”N 3 Œژ) | ||||||||||||||||||||||||||||||
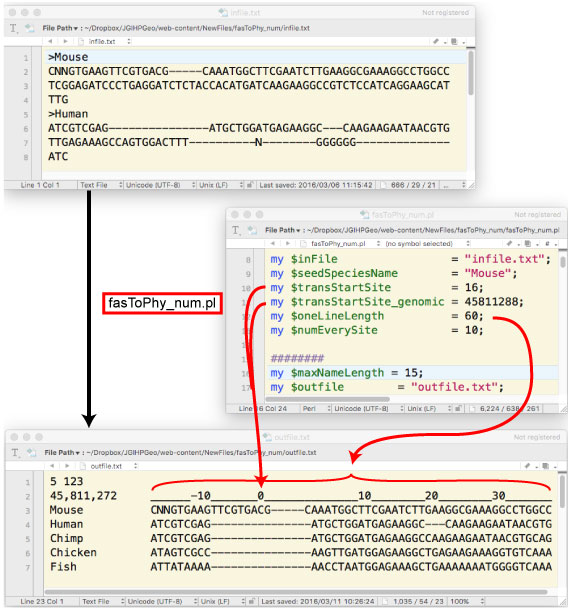 |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
motifFinder.tar.gz
my $match = "T..CACCT";
(2016 ”N 3 Œژ) |
||||||||||||||||||||||||||||||
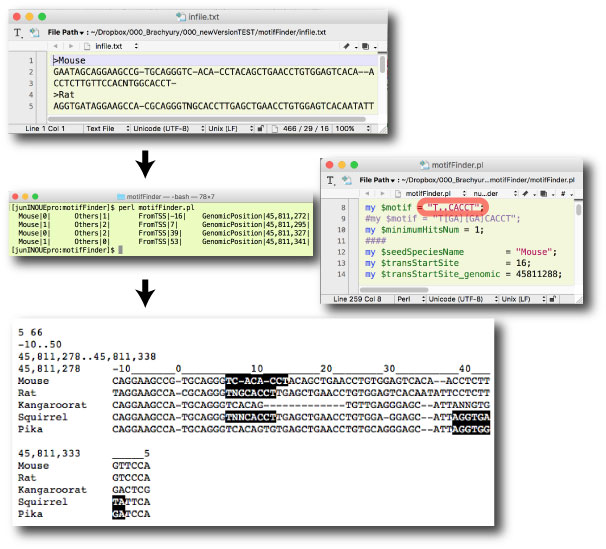 |
||||||||||||||||||||||||||||||
| motifFinder_region.tar.gz | ||||||||||||||||||||||||||||||
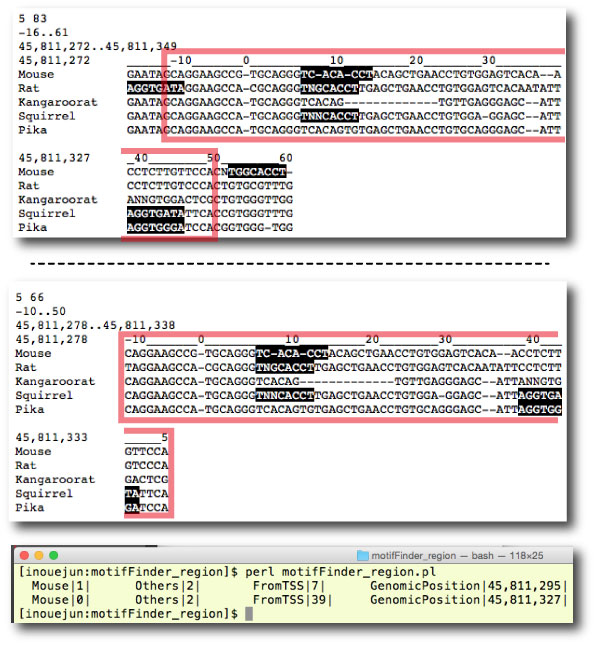 |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
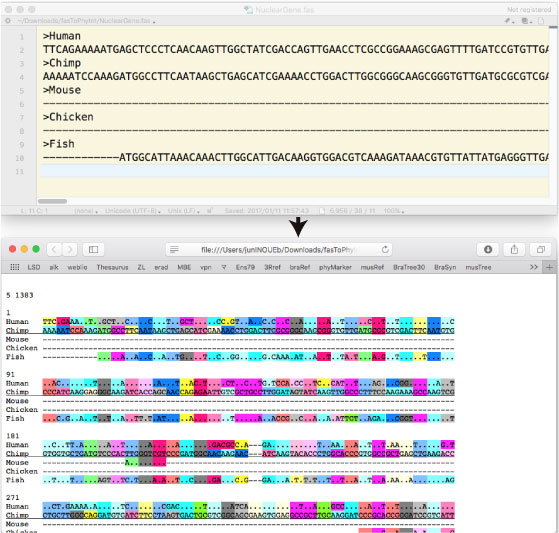
ƒAƒ‰ƒCƒپƒ“ƒgچد‚ف Fasta Œ`ژ®‚ج cDNA ‚ًپCƒRƒhƒ“•ت‚ةگF•ھ‚¯‚µپCmatch first ‚ة‚µ‚ؤŒ©‚â‚·‚‚µ‚½ƒAƒ‰ƒCƒپƒ“ƒgƒtƒ@ƒCƒ‹ (html) ‚ة•دٹ·‚·‚éƒvƒچƒOƒ‰ƒ€‚إ‚·پD |
||||||||||||||||||||||||||||||
 |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| Mesquite ‚إ•غ‘¶‚µ‚½ cDNA nexus ƒtƒHپ[ƒ}ƒbƒg‚ًˆب‰؛‚ج‚و‚¤‚ةگF•t‚« html ƒtƒ@ƒCƒ‹‚ة•دٹ·‚µ‚ـ‚·پDƒCƒ“ƒgƒچƒ“•”•ھ‚حگF‚ً•t‚¯‚¸‰؛گü‚ة‚µ‚ـ‚·پD nex2PhyInt.tar.gz |
||||||||||||||||||||||||||||||
 |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
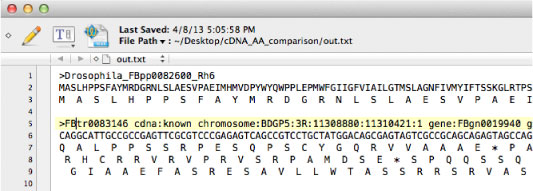
Coding sequence (DNA) ‚ً 1st, 2nd, 3rd ‚©‚çƒAƒ~ƒmژ_”z—ٌ‚ة–|–َ‚µ‚ـ‚·پD
cDNA_AA_comparison.tar.gz (2019 ”N 1 Œژ) |
||||||||||||||||||||||||||||||
 |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
“¾‚ç‚ꂽƒtƒ@ƒCƒ‹‚ً MacClade ‚إٹJ‚‚ئپCƒAƒ~ƒmژ_–|–َ‚ة‘خ‰‚µ‚½ DNA ƒAƒ‰ƒCƒپƒ“ƒg‚ًŒ©‚邱‚ئ‚ھ‚إ‚«‚ـ‚·پD |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
fasta Œ`ژ®‚جƒVپ[ƒPƒ“ƒX‚ھ 80 •¶ژڑ‚®‚ç‚¢‚إ‰üچs‚³‚ê‚ؤ‚¢‚邱‚ئ‚ھ‚ ‚è‚ـ‚·پD‚±‚ê‚ًˆêچs‚ة‚·‚éƒXƒNƒٹƒvƒg‚إ‚·پD
FasLineBreakDeleter.pl.tar.gz |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| FasDelIdenSeq_fol.tar.gz |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
ƒAƒ‰ƒCƒپƒ“ƒg‚©‚ç‰ًگح‚ة•s—v‚بƒTƒCƒg‚ًژو‚èڈœ‚«‚ـ‚·پDٹeƒTƒCƒg‚ةٹـ‚ـ‚ê‚éƒMƒƒƒbƒvگ”‚جڈمŒہ‚ًژw’肵‚ؤژو‚èڈœ‚‚±‚ئ‚ھ‚إ‚«‚ـ‚·پD
GapSiteDeleter_fol.tar.gz |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
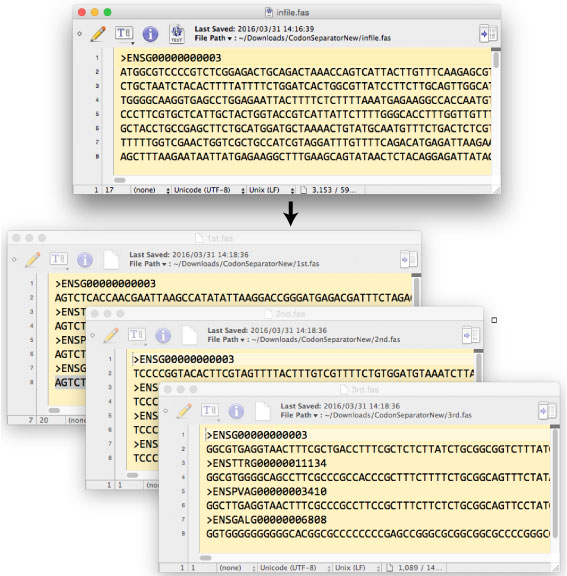
| CodonSeparatorNew.tar.gz | ||||||||||||||||||||||||||||||
 |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
1st, 2nd, 3rd ‚ئ•تپX‚ةچىگ¬‚µ‚½ fasta ƒtƒ@ƒCƒ‹‚ًپC‚à‚ئ‚جˆâ“`ژq”z—ٌ‚ة–ك‚µ‚ـ‚·پDfasta Œ`ژ®‚جƒtƒ@ƒCƒ‹‚©‚ç‰üچs‚ًژو‚èڈœ‚¢‚ؤ‚¨‚¢‚ؤ‚‚¾‚³‚¢پD—ل‘è‚حڈم‚جˆâ“`ژq”z—ٌ‚ئ“¯‚¶‚إ‚·پD Codon_combiner_fol.tar.gz |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| 3rdDeleter_fol.tar.gz |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| NucTranslaterP2N.tar.gz (2017 ”N 11 Œژ) | ||||||||||||||||||||||||||||||
| pal2nal ‚ة genetic code ‚ب‚اگف’è‚ً‚إ‚«‚邾‚¯‚ ‚ي‚¹‚ؤ‚¢‚ـ‚·پD—ل‚¦‚خپCTNC (N ‚ھٹـ‚ـ‚ê‚ؤ‚¢‚é) ‚âپ@A-A (ƒMƒƒƒbƒv‚ھٹـ‚ـ‚ê‚ؤ‚¢‚é) ‚ب‚ا‚جƒRƒhƒ“‚ح X ‚ئ‚µ‚ؤ•ش‚µ‚ـ‚·پDMacClade ‚إ‚ح‚±‚ê‚ç‚جƒRƒhƒ“‚ًپCƒAƒ~ƒmژ_–|–َ‚إ‚حƒMƒƒƒbƒv‚ئ‚µ‚ؤ•ش‚µ‚ؤ‚¢‚é‚ج‚إپC‘ٹˆل‚ھŒ©‚ç‚ê‚ـ‚·پD ƒ~ƒgƒRƒ“ƒhƒٹƒA‚جˆâ“`ژqˆأچ†‚à•t‚¯‰ء‚¦‚ـ‚µ‚½ (2017 ”N 11 Œژ)پD |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
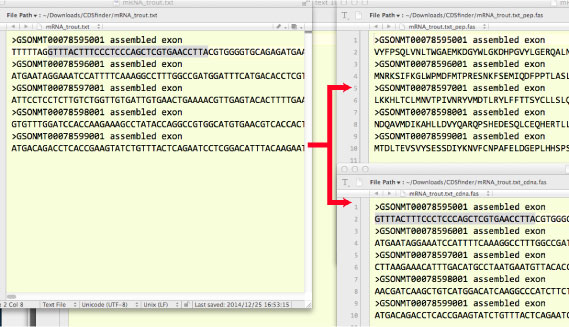
| CDSfinder.tar.gz (2014 ”N 12 Œژ) | ||||||||||||||||||||||||||||||
 |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
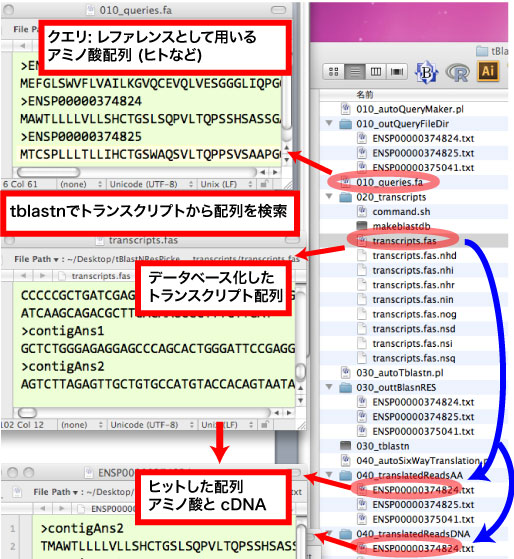 |
||||||||||||||||||||||||||||||
[inouejun:tBlastNResPicker]$ perl 010_autoQueryMaker.pl [inouejun:tBlastNResPicker]$ cd 020_transcripts/ [inouejun:tBlastNResPicker]$ perl 030_autoTblastn.pl [inouejun:tBlastNResPicker]$ perl 040_autoSixWayTranslation.pl |
||||||||||||||||||||||||||||||
| tBlastNResPicker.tar.gz (27MB) |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
ƒAƒ~ƒmژ_ƒAƒ‰ƒCƒپƒ“ƒg‚ةچ‡‚ي‚¹‚ؤپCcDNA ‚جƒAƒ‰ƒCƒپƒ“ƒg‚ًچىگ¬‚·‚éƒXƒNƒٹƒvƒg‚إ‚· [2017 ”N 12 Œژپ@‰ü’ù]پD
AAincGap.fas: ƒAƒ‰ƒCƒپƒ“ƒgچد‚ف‚جƒAƒ~ƒmژ_”z—ٌ ƒCƒ“ƒtƒ@ƒCƒ‹‚جƒMƒƒƒbƒv‚ًژو‚èڈœ‚¢‚½ڈَ‘ش‚إƒAƒ~ƒmژ_‚ئ DNA ‚إ’·‚³‚ً”نٹr‚µ‚ؤƒXƒNƒٹپ[ƒ“ƒAƒEƒg‚µ‚ؤ‚¢‚ـ‚·‚ھپC‚ ‚ـ‚è•K—v‚ب‚¢‚و‚¤‚ب‹C‚ھ‚µ‚ؤ‚«‚ـ‚µ‚½پD’·‚³‚ھˆظ‚ب‚邱‚ئ‚ھ‚و‚‚ ‚è‚ـ‚·‚ھپC‚±‚ê‚حپuA-Tپv‚ب‚ا‚حƒAƒ~ƒmژ_”z—ٌ‚إ‚ح X ‚ب‚ا‚ة‚ب‚ء‚ؤ‚¢‚邽‚ك‚¾‚ئژv‚ي‚ê‚ـ‚·پD UTRs ‚âƒ|ƒٹ A ‚ھ“ü‚ء‚ؤ‚¢‚éڈêچ‡‚حپCˆê“x pal2nal ‚إڈˆ—‚µ‚ؤ‚±‚ê‚ç‚ًژو‚èڈœ‚•K—v‚ھ‚ ‚è‚ـ‚·پD |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
DNA ‚ً‘ٹ•âچ½‚ة•دٹ·‚·‚éƒvƒچƒOƒ‰ƒ€‚إ‚·پDƒqƒ“ƒg‚¾‚¯ڈ‘‚«‚ـ‚·پDڈع‚µ‚‚ح‚±‚؟‚ç‚ًژQڈئ‚µ‚ؤ‚‚¾‚³‚¢پ@[2015 ”N 1 Œژ]پD
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| ڈd•،‚µ‚½ƒŒƒRپ[ƒh‚ًچيڈœ‚·‚éƒXƒNƒٹƒvƒg‚إ‚·پDƒnƒbƒVƒ…‚ًژg‚ء‚ؤ‚¢‚ب‚¢‚ج‚إپCƒŒƒRپ[ƒh‚جڈ‡”ش‚ً•دچX‚µ‚ـ‚¹‚ٌ [2011 ”N 5 Œژ]پD uniqSeqPicker_fol.tar.gz |
||||||||||||||||||||||||||||||
sub uniqArrayOneslf{
my $refArray = shift@_;
my @uniqArray;
foreach my $name (@$refArray) {
if(grep($_ eq "$name", @uniqArray) == 0){
push(@uniqArray,$name);
}
}
return \@uniqArray;
} |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| phySeq2fas.tar.gz (2016 ”N 3 Œژ) | ||||||||||||||||||||||||||||||
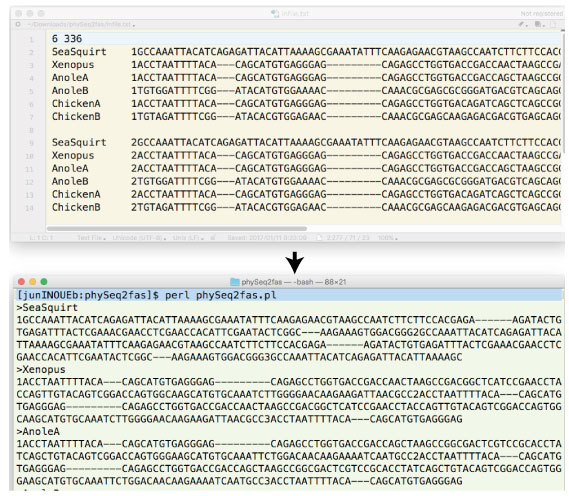 |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
NexToFas_fol.tar.gz [2010 ”N 12 Œژ] my($ref_seqHash,$ref_nameArray) = &nexReader(\@inFileCont); |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| ‚±‚ê‚àNexus Œ`ژ®‚ج”z—ٌ•”•ھ‚ةپC‰üچs‚ًٹـ‚ك‚ب‚¢‚إ‚‚¾‚³‚¢پDMatch first ‚ة‚à‘خ‰‚µ‚ؤ‚¢‚ب‚¢‚إ‚·پD NexToPhy_fol.tar.gz |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
MacClade ‚جƒRƒپƒ“ƒg—“‚ةپu#پv‚ئ‹L“ü‚µ‚½ƒTƒCƒg‚ًژو‚èڈœ‚«‚ـ‚·پDŒ‹‰ت‚ًFasta Œ`ژ®‚إ•غ‘¶‚µ‚ـ‚·پDMacClade ‚إ•غ‘¶‚µ‚½ƒtƒ@ƒCƒ‹‚ج‰üچsƒRپ[ƒh‚ح Mac Œ`ژ®‚إ‚·‚ھپCژ©“®“I‚ة Unix Œ`ژ®‚ة•دٹ·‚µ‚ـ‚·پD
NexToPhy_fol.tar.gz |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
•،گ”‚جژي‚©‚瓾‚ç‚ꂽ‘ٹ“¯ˆâ“`ژq‚ج cDNA ”z—ٌ‚ھ•غ‘¶‚³‚ꂽ GenBank format (ˆê‚آ‚ة‚آ‚ب‚ھ‚ء‚½ƒtƒ@ƒCƒ‹) ‚©‚çپCƒAƒ~ƒmژ_”z—ٌ‚ئ‰–ٹî”z—ٌ‚ً’ٹڈo‚µ‚ؤƒAƒ‰ƒCƒپƒ“ƒg‚ًچs‚¢‚ـ‚·پDGenBank format ‚ح‰½ژي“ü‚ء‚ؤ‚¢‚ؤ‚à‘هڈن•v‚إ‚·‚ھپCˆâ“`ژq‚ح 1 ‚آ‚¾‚¯‚إ‚·پDˆâ“`ˆأچ†‚جٹضŒWڈمپCƒ~ƒgƒRƒ“ƒhƒٹƒAˆâ“`ژq‚ة‚ح‘خ‰‚µ‚ؤ‚¢‚ـ‚¹‚ٌ (pal2nal ‚حƒ~ƒgƒRƒ“ƒhƒٹƒAˆâ“`ژq‚إ‚حگ³ڈي‚ةچى“®‚µ‚ب‚¢‚½‚ك‚إ‚·) [2010 ”N 10 Œژ]پD
ڈم‹L‚جƒRƒ}ƒ“ƒh‚ة‚و‚ء‚ؤپCmafft (ƒCƒ“ƒXƒgپ[ƒ‹‚·‚é•K—v‚ھ‚ ‚è‚ـ‚·)پCpal2nal, FasToNex_cDNA.pl ‚ھژ©“®“I‚ةچى“®‚µ‚ؤپCƒAƒ~ƒmژ_‚جƒAƒ‰ƒCƒپƒ“ƒg‚ھچs‚ي‚êپCcDNA ‚جƒAƒ‰ƒCƒپƒ“ƒg‚à“¾‚ç‚ê‚ـ‚·پD |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
mt ƒQƒmƒ€”z—ٌ‚ھ•غ‘¶‚³‚ꂽ GenBank format (•،گ”‚جژي) ‚©‚çƒAƒ~ƒmژ_”z—ٌ‚ً’ٹڈo‚·‚éƒXƒNƒٹƒvƒg‚إ‚·پD‰–ٹî”z—ٌ‚ح“¯‚¶ GenBank format ‚ً GenBankStrip.pl ‚ة‚و‚ء‚ؤڈˆ—‚·‚ê‚خ“¾‚ç‚ê‚ـ‚·پDOTU –¼‚ةƒXƒyپ[ƒX‚ھ“ü‚ء‚ؤ‚¢‚é‚ئ MacClade ‚إ“ا‚فچ‚ق‚ئ‚«‚ب‚ا‚ة–â‘è‚ة‚ب‚é‚ج‚إپCEditor ‚ًژg‚ء‚ؤ•دچX‚·‚é•K—v‚ھ‚ ‚è‚ـ‚· [2010 ”N 10 Œژ]پD aaSeqRetFromGBK_fol.tar.gz |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
P distance (•دˆظƒTƒCƒg/‘S’·) ‚ًŒvژZ‚µ‚ـ‚·پDpaup ‚و‚èژلٹ±’ل‚¢’l‚ھڈo‚ـ‚·پD——R‚ح’²‚ׂؤ‚¢‚é‚ئ‚±‚ë‚إ‚·پD
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
jason2seqnece.tar.gz |
||||||||||||||||||||||||||||||
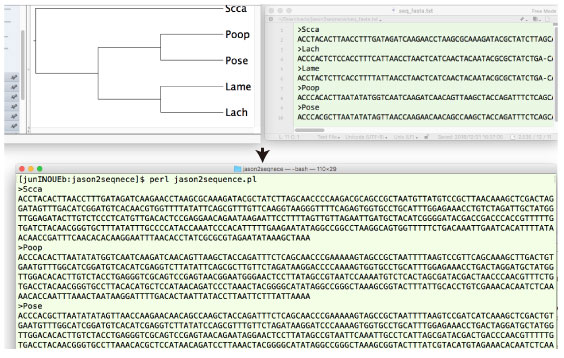 |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| sequence file ‚ة•ہ‚ׂç‚ꂽڈ‡”ش‚ً”شچ†‚ئ‚µ‚ؤپCtree file ‚جژي–¼‚ً”شچ†‚ة’u‚«ٹ·‚¦‚ـ‚·پD SpNameToNum_fol.tar.gz |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
my $tree = '(4,(3,(2,1)))'; |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
newick Œ`ژ®‚ً“ا‚ٌ‚إپCƒNƒŒپ[ƒh‚ةٹـ‚ـ‚ê‚é leaf ‚ًƒNƒŒپ[ƒh‚ج‘ه‚«‚¢ڈ‡‚ةڈ‘‚«ڈo‚·پD |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
“ٌ‚آ‚جŒn“ژ÷‚ةٹـ‚ـ‚ê‚é OTU چ\گ¬‚ح“¯‚¶‚ة‚µ‚ؤ‚‚¾‚³‚¢پDˆâ“`ژq‚جŒn“ژ÷ (”ن‚ׂé‘خڈغ) ‚ج OTU –¼‚جچإڈ‰‚حپCژي‚جŒn“ژ÷‚إ—p‚¢‚½ OTU –¼‚ة‚µ‚ؤپCپu_پv(ƒAƒ“ƒ_پ[ƒoپ[) ‚ً‹²‚ٌ‚إˆâ“`ژq–¼‚ب‚ا‚ًڈ‘‚‚و‚¤‚ة‚µ‚ؤ‚‚¾‚³‚¢ [2012 ”N 3 Œژ]پD |
||||||||||||||||||||||||||||||
[inouejun:treeComparison]$ perl treeComparison.pl |
||||||||||||||||||||||||||||||
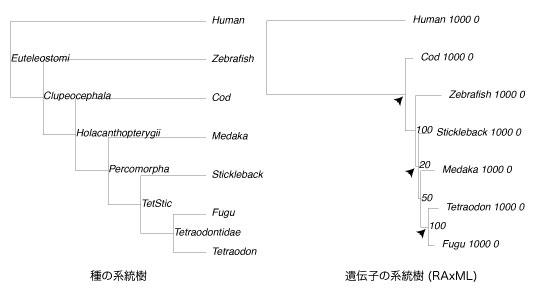 |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
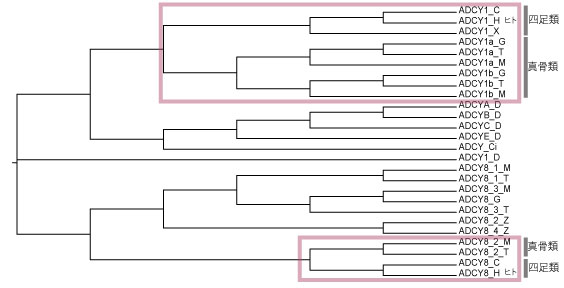
Human (_H)‚ئ teleosts ('_Z','_M','_S','_T') ‚ج—¼•û‚ھٹـ‚ـ‚ê‚éچإڈ¬‚جƒNƒŒپ[ƒh‚ً‘I‚ٌ‚إ (‰؛گ})پCleaf ‚ًڈ‘‚«ڈo‚µ‚ـ‚·پD
(ˆب‰؛‚ح–â‘è‚ھ‚ ‚é‚ج‚إژg‚ي‚ب‚¢‚إ‚‚¾‚³‚¢)
|
||||||||||||||||||||||||||||||
 |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
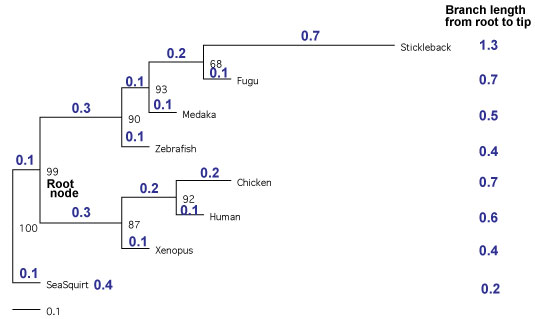
ٹOŒQ 1 ژي‚جژ÷’·•t‚« rooted tree ‚ً‰ًگح‚µ‚ؤپCچھٹ²‚©‚ç––’[‚ـ‚إ‚جژ}’·‚ًژي‚²‚ئ‚ةŒvژZ‚µ‚ـ‚· [2011 ”N 11 Œژ]پD |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
 |
||||||||||||||||||||||||||||||
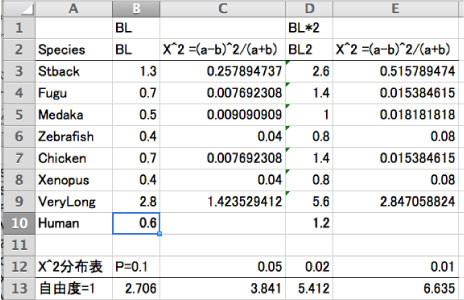
| ژv‚¤‚و‚¤‚بŒ‹‰ت‚ھ“¾‚ç‚ê‚ـ‚¹‚ٌ‚إ‚µ‚½‚ھپC“ْ–{Œê”إ Yang (2006) P 217 ‚ة‚ ‚éپ@Fitch (1976) ‚ج•û–@‚إ‘ٹ‘خ‘¬“xƒeƒXƒg‚ًچs‚ء‚½‚ج‚إ‰ًگà‚µ‚ـ‚·پD—ل‚ئ‚µ‚ؤپCڈم‚جگ}‚ة‚ ‚éژ}’·•t‚«Œn“ژ÷‚ًژg‚ء‚ؤ‚¢‚ـ‚·پD‘¼‚ة‚à‘ٹ‘خ‘¬“xƒeƒXƒg‚ھڈذ‰î‚³‚ê‚ؤ‚¢‚ـ‚·‚ھپC•Wڈ€Œëچ·‚ً”z—ٌ‚ً‰ًگح‚µ‚ؤ‹پ‚ك‚½‚è‚·‚é•K—v‚ھ‚ ‚èپCperl ‚¾‚¯‚إٹب’P‚ة‚إ‚«‚»‚¤‚ة‚ب‚¢‚ج‚إژژ‚µ‚ؤ‚¢‚ـ‚¹‚ٌپD ‚±‚±‚إ‚ح Human ‚ئ‘¼ژي‚جٹش‚إژ}’·‚ً”نٹr‚µپCƒJƒC“ٌڈو“Kچ‡“xŒں’è‚ة‚و‚ء‚ؤ‘¼ژي‚جژ}’·‚ھٹü‹p‚إ‚«‚é‚© (‚ ‚蓾‚ب‚¢’·‚³‚©) ‚ًŒں’肵‚ؤ‚¢‚ـ‚·پDYang (2006) ‚إ‚حپ@5 چs–ع‚ة‚¨‚¯‚éژ}’·‚جگ„’è‚ھپCBL ‚ً 2 ”{‚µ‚½‚à‚ج‚ة‚ب‚ء‚ؤ‚µ‚ـ‚ء‚ؤ‚¢‚é‚و‚¤‚ة‚àژَ‚¯ژو‚ê‚é‚ج‚إپCBL ‚ئ BL*2 ‚ج—¼•û‚ًŒvژZ‚µ‚ـ‚µ‚½پD‚±‚ج–{‚ة‚و‚é‚ئپCƒJƒC“ٌڈو“Kچ‡“xŒں’è‚حژ©—R“x 1 ‚إچs‚¤‚و‚¤‚إ‚·پD ڈم‚جگ}‚ج‚و‚¤‚ةپC–{—ˆ‚ب‚ç‚خ Stback ‚ھٹü‹p‚³‚ê‚ؤ‚ظ‚µ‚©‚ء‚½‚ج‚إ‚·‚ھپCƒJƒC“ٌڈو (X^2) ‚ج’l‚حپCBL2 ‚إ‚ ‚ء‚ؤ‚à 0.5157.. ‚ئپC—Lˆسگ…ڈ€ 0.1 (P=0.1) ‚ج’l 2.706 (ƒJƒC“ٌڈو•ھ•z•\‚و‚è) ‚ة‰“‚‹y‚خ‚ب‚¢‚إ‚·پD ژژ‚µ‚ة BL ‚ً 2.8 ‚ة‚·‚é‚ئپC‚و‚¤‚â‚ BL2 ‚إŒvژZ‚µ‚½ڈêچ‡‚ح P=0.1 ‚إٹü‹p‚إ‚«‚ـ‚µ‚½پD‚±‚جڈêچ‡پCHuman ‚ج–ٌ 4.7 ”{‚ة‚à‚ب‚ء‚ؤ‚µ‚ـ‚¢‚ـ‚·پD‚±‚ج•û–@‚ھ‚¨‚©‚µ‚¢‚ج‚©پCژ„‚ج‰ًژك‚ھٹشˆل‚ء‚ؤ‚¢‚é‚ج‚©پCچ،‚ج‚ئ‚±‚ë‰ًŒˆ‚إ‚«‚ؤ‚¢‚ـ‚¹‚ٌپDژہچغ‚جƒfپ[ƒ^‚ًژg‚ء‚ؤ‚à“¯‚¶‚و‚¤‚بŒ‹‰ت‚ھ“¾‚ç‚ê‚ؤ‚µ‚ـ‚¢‚ـ‚µ‚½ [2011 ”N 11 Œژ]پD =>•½‹د’l (ژ}’·ٹش) ‚©‚ç‚جٹu‚½‚è‚ً‘خڈغ‚ئ‚µ‚½‘ٹ‘خ‘¬“xƒeƒXƒg‚إ‚ ‚ê‚خپCLINTREE ‚ًژg‚¤‚±‚ئ‚à‰آ”\‚إ‚·پD‚½‚¾پCژ„‚جٹ´گG‚إ‚حپCLINTREE ‚ح‹t‚ةگ¸“x‚ھچ‚‚·‚¬‚é‹C‚ھ‚µ‚ـ‚·پD |
||||||||||||||||||||||||||||||
 |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
@INC (ƒCƒ“ƒN) ‚ح Perl ‚ھ‚ ‚ç‚©‚¶‚ك’è‹`‚µ‚ؤ‚¢‚é”z—ٌ•دگ”‚إ‚·پD‚±‚±‚ة require ‚ب‚ا‚جƒRƒ}ƒ“ƒh‚ھƒ‰ƒCƒuƒ‰ƒٹ‚ً’T‚µ‚ةچs‚ƒfƒBƒŒƒNƒgƒٹ‚جˆê——‚ھ•غژ‚³‚ê‚ؤ‚¢‚ـ‚·پD
‚ج‚و‚¤‚بƒGƒ‰پ[ƒپƒbƒZپ[ƒW‚ھڈo‚邱‚ئ‚ھ‚ ‚è‚ـ‚·پD‚±‚ê‚ح micoode.pl ‚ھŒ©‚آ‚©‚ç‚ب‚¢‚ئ‚¢‚¤ˆس–،‚إ‚·پD‚±‚جڈêچ‡‚حپC
‚ئ‚¢‚¤ƒRƒ}ƒ“ƒh‚ً‘إ‚آ‚±‚ئ‚إپCperl ‚ھ micode.pl ‚ً’T‚µ‚ؤ‚¢‚éƒfƒBƒŒƒNƒgƒٹ‚جƒAƒhƒŒƒX‚ًٹm”F‚·‚邱‚ئ‚ھ‚إ‚«‚ـ‚·پD |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| ƒ}ƒbƒ`‰‰ژZژq‚ج—ûڈKƒvƒچƒOƒ‰ƒ€‚إ‚·پD | ||||||||||||||||||||||||||||||
ƒAƒEƒgƒvƒbƒg‚إ‚·پD
|
||||||||||||||||||||||||||||||
| پuگV”إPerlŒ¾ŒêƒvƒچƒOƒ‰ƒ~ƒ“ƒOƒŒƒbƒXƒ““ü–ه•زپv ‚جƒTƒ“ƒvƒ‹ƒvƒچƒOƒ‰ƒ€‚ًژQچl‚ة‚µ‚ـ‚µ‚½پD‚±‚؟‚ç‚إ‚·پD |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
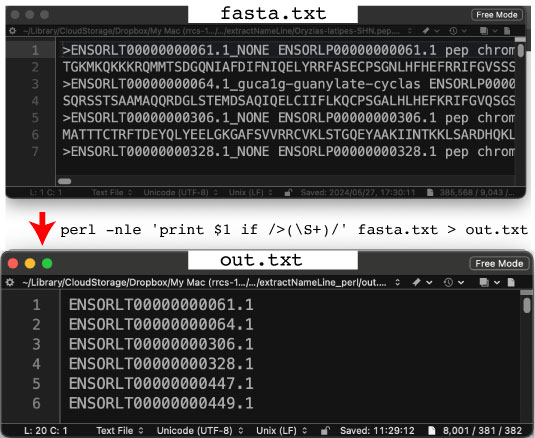
| extractNameLine_perl.tar.gz (2024 ”N 7 Œژ) | ||||||||||||||||||||||||||||||
 |
||||||||||||||||||||||||||||||
-e: ˆّ—p•„‚إˆح‚ـ‚ꂽ 'perl $1 if />(\S+)/' •”•ھ‚ھƒXƒNƒٹƒvƒg‚إ‚ ‚邱‚ئ‚ًژ¦‚· (link)پB
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| پu‰يâؤ‚ةڈو‚éگl’S‚®گl‚»‚ج‚ـ‚½‘گèـ‚ًچى‚éگlپv by A.T. |
||||||||||||||||||||||||||||||