|
||||
| 2026 年 5 月 28 日 改訂 | ||||
|
||||
インストールから学ぶには、以下のページを参照してください。
Ubuntu 環境の Python に従いました。 ビルドライブラリのインストール
ソースコードのダウンロード 非公式 Python ダウンロードリンクから Python-3.12.1.tar.xz をダウンロードしました。その後、以下で解凍しました。
ビルド
ビルドしたコマンドは /usr/local/bin にインストールされます。python3 あるいは python3.12 コマンドで起動します。 ソースからビルドする: CentOS7 下準備 いくつかの Python モジュールは、外部のパッケージに依存しているそうです。これら外部パッケージのインストールは、Python のビルドを行う前に行う必要があります。以下をインストールします。
Python のインストール
|
||||
|
||||
Ubuntu: System wide に numpy と pandas をインストールする こちらのサイトにしたがって以下を行うと、/usr/bin にある python3 (python3.12.3) にインストールされました。インストールの確認は、以下、「numpy がインストール済みか確認」を参照してください。
Ubuntu では、Sisteym wide に、すなわち、sudo によって、サーバーを利用する全てのユーザーが numpy と pandas を利用できるようなインストールを行うには、pip ではなく、apt-get install python3-xxxx が良いそうです。
ちなみに、以前は、pip を使って以下を実行していましたが、ubuntu では上記の方法は推奨されていません。
さらに、Ubuntu をインストールしたら自動的に入っている python3 の場合は、上記 sudo コマンドでは numpy と pandas をインストールできませんでした。 numpy がインストール済みか確認
|
||||
エラーメッセージ
|
||||
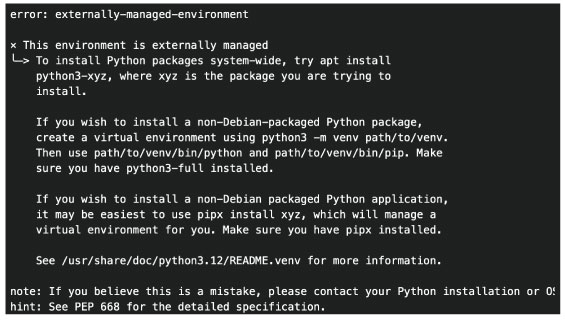
pip でインストールできない場合は、上のようなエラーが出ます。こちらを参照してください。 (2014 年 10 月)。
|
||||
|
||||
#!/usr/bin/env python test.py という名前にして保存してください.
によって実行可能権をスクリプトにつけておきます.env は which と似たようなもので,python を自動的に探し出します. [inouejun:scripts]$ test.py |
||||
|
||||
>>> A=[1,2,3] |
||||
|
||||
with open("infile") as fileobject: |
||||
|
||||
#!/usr/bin/env python3
from collections import OrderedDict
def readFasta_dict(filename):
seq_dict = OrderedDict()
with open(filename, "r") as infile:
name = None # 変数 name を初期化
for line in infile:
line = line.strip()
if not line:
continue # 空行をスキップ
if line.startswith(">"):
name = line
seq_dict[name] = ""
elif name:
seq_dict[name] += line
else:
raise ValueError("ERROR: ヘッダーの前に配列があります。")
return seq_dict
# 正しいファイル名に修正
infile_name = "fasta.txt"
seq_dict = readFasta_dict(infile_name)
for name, seq in seq_dict.items():
print(name)
print(seq)
|
||||
|
re.compile を使った方法は,こちらを参照. readFasta.tar.gz 2025 年 5 月: copilot により改訂 |
||||
|
||||
 |
||||
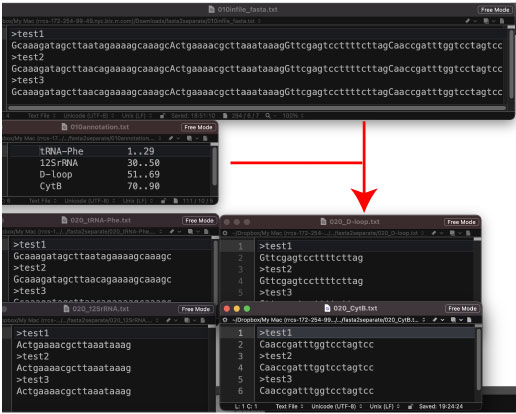
fasta2separate.tar.gz (2022 年 2 月)
|
||||
|
||||
 |
||||
| phyCodon2Block.tar.gz (2018 年 2 月) |
||||
|
||||
 |
||||
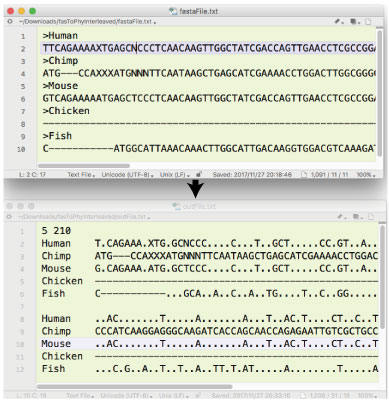
| fasToPhyInterleaved.tar.gz (2017 年 11 月) |
||||
|
||||
 |
||||
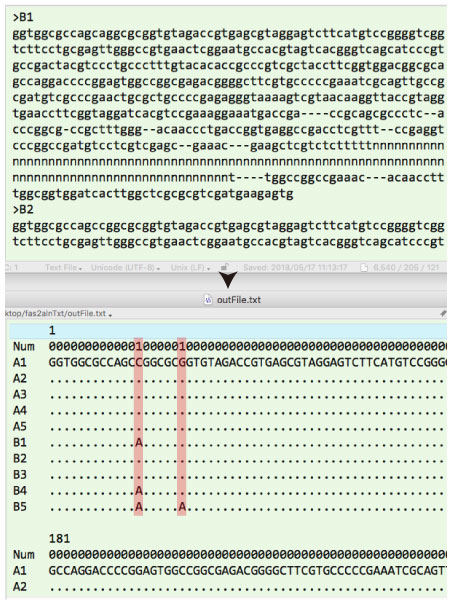
| fas2alnTxt.tar.gz (2018 年 5 月) |
||||
|
||||
 |
||||
files_changeWord.zip (2026 年 5 月)
|
||||
|
||||
 |
||||
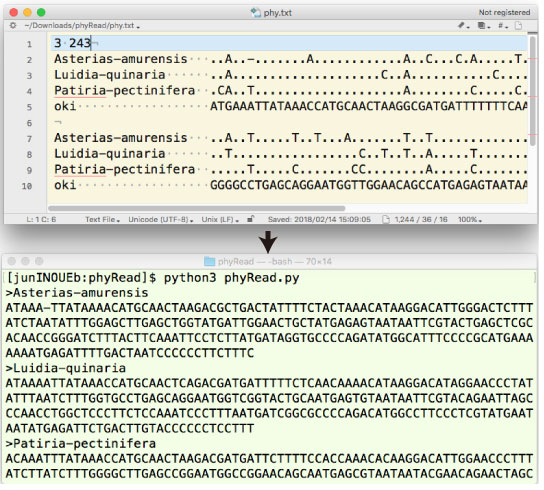
| phyRead.tar.gz (2018 年 2 月) |
||||
|
||||
def translation(dna): |
||||
| translation.tar.gz (2018 年 2 月) |
||||
|
||||
 |
||||
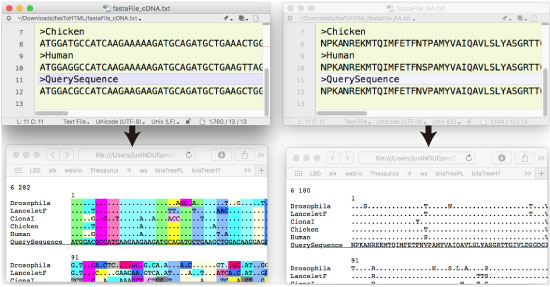
fasToHTML.tar.gz (2017 年 11 月) |
||||
|
||||
line = "gi|1169025|COX1_CAEEL" line = ">gi|1169025|COX1_CAEEL" import re line = "ATATGTGTGAAA" import re line = "ATGTGTGTGAAAAA" import re import re [junINOUEpro:Downloads]$ python3 test.py import re [junINOUEpro:Downloads]$ python3 test2.py |
||||
|
||||
rec = {
import re
protID = "ENSACAP00000000002"
recs = {
">ENSACAP00000000002 pep1" : "ATGCTG",
">ENSACAP00000000003 pep" : "AGGCTG",
">ENSACAP00000000002 pep2" : "ACGCTG",
">ENSACAP00000000005 pep" : "AAGCTG",
">ENSACAP00000000006 pep" : "ATACTG"
}
key = ">" + protID + " \w*"
hitNames = [name for name in recs.keys() if re.search(key,name)]
if len(hitNames) > 1:
print("More than 2 hits.")
print(hitNames)
elif len(hitNames) == 1:
print("1 hit.")
print(hitNames)
else:
print("Not hit.")
|
||||
|
||||
import re [junINOUEpro:Downloads]$ python3 test.py import re
string = ">FBpp0082536 gene:xxx"
rr = re.sub(r">([^ ]+) (gene:.*$)", r">\1 A:\1 ", string)
print("rr: ", rr)
junINOUEpro:Downloads]$ python test.py rr: >FBpp0082536 A:FBpp0082536 line = "Anguilla japonica" [junINOUEpro:Downloads]$ python3 test2.py |
||||
|
||||
import re [junINOUEpro:Downloads]$ python3 test2.py import re [junINOUEpro:Downloads]$ python3 test2.py |
||||
|
||||
|
||||
| source: sys_argv.py |
||||
|
||||
 |
||||
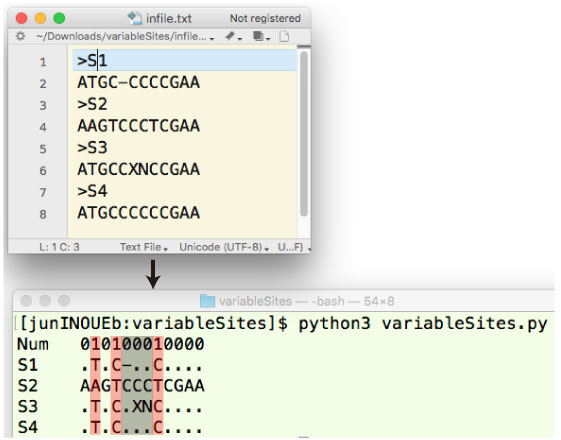
variableSites.tar.gz (2019 年 11 月) |
||||
|
||||
 |
||||
| retrieve_gene.tar.gz | ||||
(2020 年 7 月) |
||||
|
||||
import os |
||||
|
||||
import subprocess
subprocess.call("date", shell=True)
|
||||
|
||||
import subprocess bytes 型と str 型の変換についてはこちら. |
||||
|
||||
import sys argument1 = sys.argv[1] argument2 = int(sys.argv[2]) |
||||
|
||||
[junINOUEpro:compare2dirs]$ python3 missFile.py |
||||
|
||||
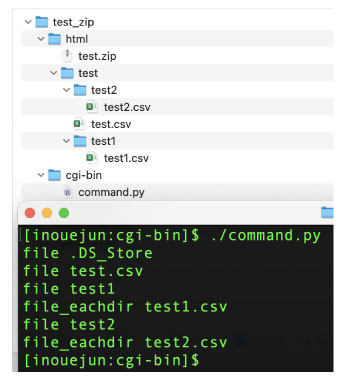 |
||||
#!/usr/local/bin/python3
import os, zipfile
myzip = zipfile.ZipFile('../html/test.zip','w')
files = os.listdir(path="../html/test/")
for file in files:
print("file", file)
myzip.write("../html/test/" + file, file)
if os.path.isdir("../html/test/" + file):
files_eachdir = os.listdir(path="../html/test/" + file)
for file_eachdir in files_eachdir:
print("file_eachdir", file_eachdir)
myzip.write("../html/test/" + file + "/" \
+ file_eachdir, file + "/" + file_eachdir)
|
||||
|
(2022 年 7 月)
|
||||
import zipfile import glob import os |
||||
|
||||
|
||||
|
||||
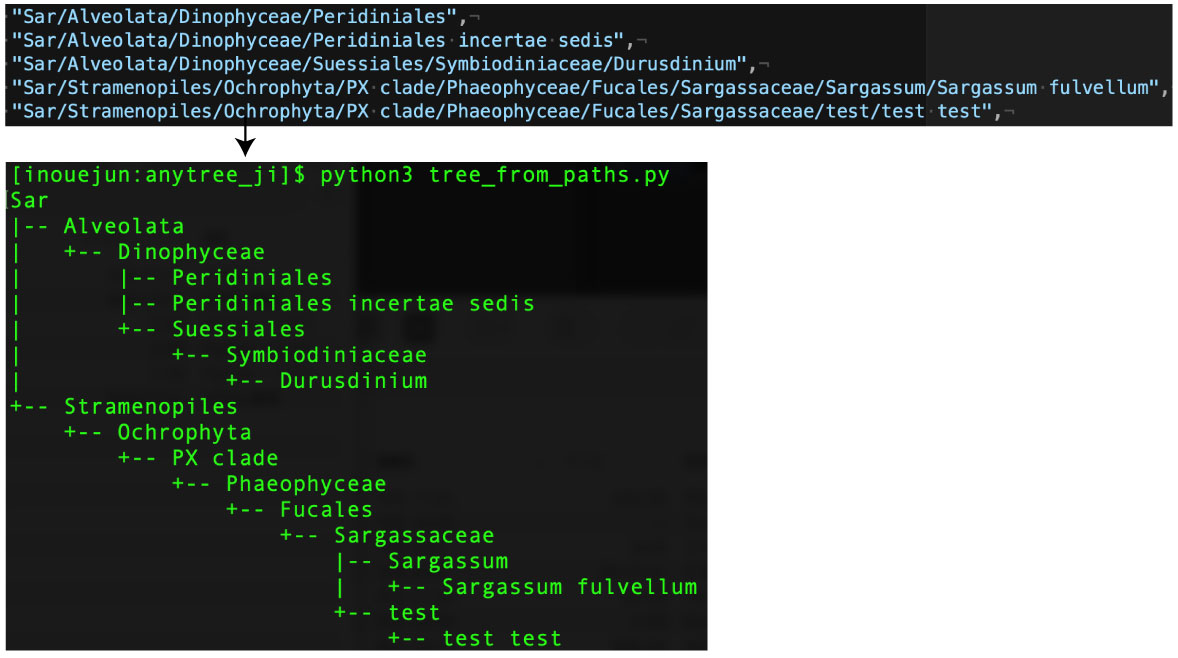
入力は Sar/Alveolata/Dinophyceae/Peridiniales のように階層をスラッシュ区切り |
||||
|
||||
import re
tree = '(4,(3,(2,1)));'
#tree = '(4,((3,5),(2,1)));'
#tree = '(((4,6),7),(3,5),(2,1));'
cladeReg = "\(([^\(\)]+)\)"
clades = []
while re.search(cladeReg, tree):
match = re.search(cladeReg, tree)
clades.append(match.group())
tree = re.sub(cladeReg, r"\1", tree, 1)
for clade in clades:
print(clade)
[junINOUEpro:tree]$ python3 cladeCollect.py
|
||||
|
||||
 |
||||
sort_seq_by_tree.tar.gz |
||||
|
||||
コマンドラインで指定する
その後,b で breakpoint を設定し,c でその場所に移動します.コメントアウトした行には breakpoint を打てないです.こちらを参照してください. |
||||
プログラムに書き込む |
||||
import pdb
line = "gi|1169025|COX1_CAEEL"
pdb.set_trace()
if "COX1" in line:
print("Found") |
||||
[junINOUEpro:Downloads]$ python3 test.py > /Users/junINOUEpro/Downloads/test.py(5)<module>() -> if "COX1" in line: (Pdb) l 1 import pdb 2 3 line = "gi|1169025|COX1_CAEEL" 4 pdb.set_trace() 5 -> if "COX1" in line: 6 print("Found") [EOF] (Pdb) p line 'gi|1169025|COX1_CAEEL' (Pdb) n > /Users/junINOUEpro/Downloads/test.py(6)<module>() -> print("Found") (Pdb) n Found --Return-- > /Users/junINOUEpro/Downloads/test.py(6)<module>()->None -> print("Found") |
||||
|
||||
PypeR_NJ.tar.gz
こちらを参照しました (2017 年 12 月). |
||||
|
||||
Practical computing for biologists
Advanced Python for Biologists
Biology Meets Programming: Bioinformatics for Beginners
Workign With Tree Data Structures
Bioinformatics Programming Using Python
Python で大量のファイルを並列で速く読み込む (2019 年 6 月) Python for biologists を用いた演習 (2019 年 6 月) Pythonプログラムを稼働させるのにおすすめのVPSサーバーランキング
|
||||
|
|