|
|
|
|
Ensembl はゲノムプロジェクトが行われている生物のデータだけに特化して,ゲノムアノテーションを詳細に行ったデータベースです.私の印象では,NCBI は遺伝子配列の蓄積に力を入れている一方で,Ensembl はゲノム情報の質向上を目指しているようです.実際,NCBI では cDNA 配列に対応するアミノ酸配列が登録されていない場合がよくありますが,Ensembl では完全に対応した配列が公開されています.ここ数年で急激に数が増えると思っていましたが,予想に反して最近は種数が増加しません.
Ensembl では,配列間でオーソロガスな比較が行えるような検討がなされています.こちらをご覧下さい.しかし,Ensembl のオーソログ判定は,後生動物全体を含む巨大な遺伝子系統樹に基づいているため,参考程度に考えたほうが良いと思います.
脊椎動物以外のデータは,異なるサイトにまとめられています.
EnsemblMetazoa, EnsemblPlants, EnsemblFungi, EnsemblProtists, EnsemblBacteria
|
|
| 脊椎動物については, こちらをご覧下さい.わかりにくいですが,一番下に無脊椎動物 (EnsemblMetazoa) や植物 (EnsemblPlants),菌類 (EnsemblFungi) などへのリンクが貼られています. |
 |
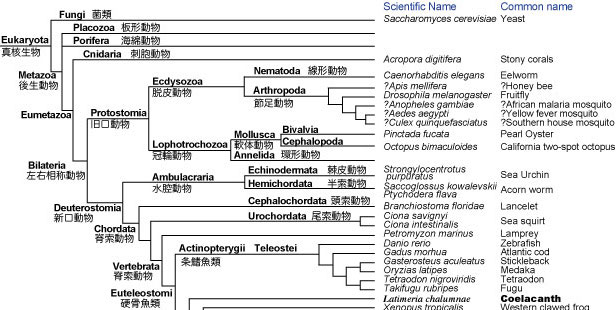
Ensembl のデータを中心として,ゲノムデータが公開されている新口動物を以下にまとめています (2016 年 4 月).Ensembl ID と外部へのリンクはこちらにまとめました.
ある種の一般的な名前や分類を調べる際は,NCBI が提供している分類のサイト (こちら) を使うと便利です.いくつかの種が含まれる分類群名を調べる場合は,こちらです. |
DatabaseGenomeSpeciesTree.pdf
|
|
Orthology information
Ensembl の最も大きな特徴の一つとして,種間で遺伝子配列を比較する場合に,その遺伝子が orthologous な関係にあるのか,paralogous な関係にあるのかを,orthology information として提供している点です.例えば,Ensembl から得られた核遺伝子配列に基づいて種の系統樹を構築する場合,この情報を用いる必要があります.私は BioMart を使って orthology information を得ています.下にあるコラムを参照してください.
Ensembl では,orthology information を得るために,遺伝子ごとに系統樹を推定して,これを種の系統樹 (これまでの形態や分子などの情報から広く受け入れられている系統関係) と比較しています.比較する遺伝子が種の分岐に起源していれば orthologous,遺伝子重複によって分岐していれば paralogous としています.遺伝子の系統樹は,それぞれの遺伝子にコードされている最長のアミノ酸配列を系統解析して得ています.近縁種の場合は PAML も使っているようです.
注意:Ensembl は真核生物全体を含む遺伝子系統樹を推定して orthology 判定を行っているので,厳密な orthology 情報ではない点に注意してください.
リンク1,リンク2
Vilella, AJ, J Severin, A Ureta-Vidal, L Heng, R Durbin, E Birney. 2009. EnsemblCompara GeneTrees: Complete, duplication-aware phylogenetic trees in vertebrates. Genetical Research 19:327–335.
Gene tree
Ensembl にコンタクトして得た情報です.Orthology information を得るのに用いられた gene tree はこちらから得ることができます.この ftp サイトは FTP Download というページにある table の上からリンクが張られています.Multi-species data の table にある Comparative genomics の行にある EMF をクリックしてください.
Compara.65.protein_trees.nhx.emf.gz に NHX フォーマットの tree が保存されています.ただし tree は巨大なので,tree を見るには工夫が必要です.NHX フォーマットの tree は Notung で見ることができます.詳細は README ファイルを見てください [2011 年 12 月].
|
|
Orthology information
BioMart を使うと,Orthologous な遺伝子を示す table などをわりと簡単に作成することができます.
Ensembl による Video Tutorials,
統合 TV,
例として,Human と Tarsius の orthology information を得る手順を説明します.
1.
BioMart に入り,CHOSE DATABAES から Ensembl 56 (最新版) を選びます.CHOOSE DATASET から Tarsius syrichta を選びます.
2.
Filters は以下を選びます.
'Orthologous Human Genes only'
Gene type は 'protein_coding' を選んでいますが,得られる結果 (遺伝子数) は一緒でした.
3.
Attribute は
'Ensembl Gene ID', 'Ensembl Human ID', 'orthology type'.
を選びます.
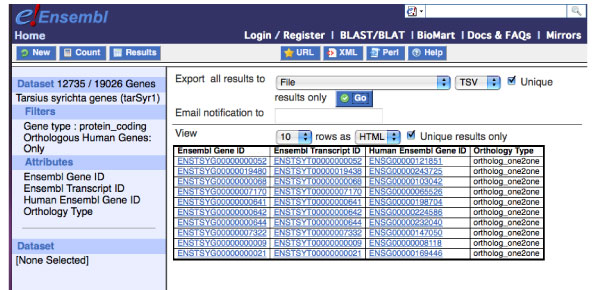
Orthology にも複数の種類があります.一つの遺伝子に対して一つだけの遺伝子が対応しているものが系統解析には都合が良いすが,実際には複数の遺伝子が対応していることも多いです.Ensembl は,最尤法を用いて真核生物 (Eukaryota) 全体の遺伝子系統樹を推定することによって,orthologous gene を大まかに判定しています.こちらをご覧下さい.
得られる遺伝子数は,Ensembl のバージョンによって異なります.Ensembl ではヒトの遺伝子数がバージョンアップごとに多くなっています.ヒト以外の種ではほとんど変化しないようです.
Ensembl: Collecting Orthologs
系統解析に利用可能な相同遺伝子グループを集めるスクリプト集を作りました. こちらです.BioMart と fasta 形式でダウンロードできる種別データベースを用いています [2012 年 12 月].
ミトコンドリアゲノム・タンパク質遺伝子配列を得る
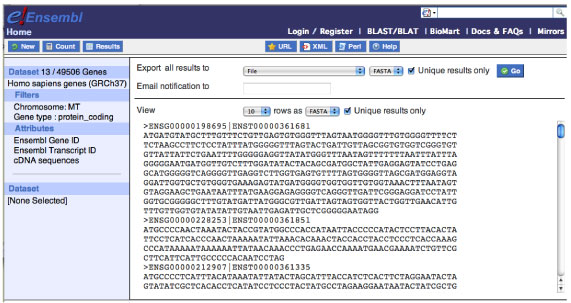
BioMart を使って,mtDNA の配列を得ることもできます.ただ, NCBI のサイトから得た方が,効率は遥かに良いように思えます.
Homo sapiens が例です.
1.
Database を Ensembl 56 (あるいは最新のもの) にする.
Dataset は Homo sapiens を選ぶ.
2.
Filters を MT (chromosome) にする.
3.
Atrributes を cDNA にする.
|
|
注意:ダウンロードした Ensembl のゲノムデータ (こちら) に,ミトコンドリアゲノムにコードされている遺伝子も入っています.2010 年ぐらいに help desk に聞いたところ,これらダウンロードデータにミトコンドリアゲノムのデータは入っていないと言いっていました,2015 年には四足類や真骨類のデータにこれらミトコンドリア遺伝子が入っていました.
|
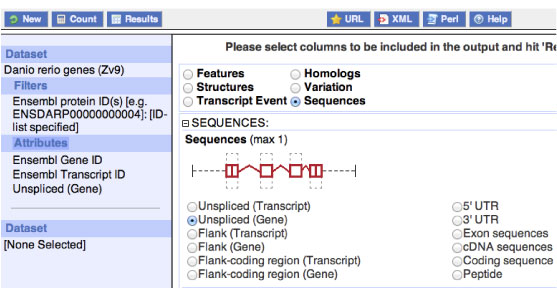
スプライシングされていない遺伝子配列を得る
BioMart を使って,スプライシングされていない遺伝子配列を得ることができます.Attributes で SEQUENCES > Unspliced (Gene) を選びます.大量データをダウンロードするには時間がかかりますが,スクリプトを書く手間などを考えると,BioMart の方がずっと楽だと思います. |
|
|
Ensembl で保存されているゲノムデータは,Human,Mouse,Zebrafish で極端に大きいです:
Homo_sapiens.GRCh38.dna_rm.toplevel.fa
57,87 GB (57,866,332,009 byte)
4.5 Gbp (4,537,931,177 bp)
Pan_troglodytes.Pan_tro_3.0.dna_rm.toplevel.fa
3.3 GB (3,288,623,126 byte)
3.4 Gbp (3,385,800,935 bp)
Mus_musculus.GRCm38.dna_rm.toplevel.fa
12.76 GB (12,760,450,175 byte)
3.4 Gbp (3,486,944,526 bp)
Canis_familiaris.CanFam3.1.dna_rm.toplevel.fa
2.4 GB(2,451,412,943 byte)
2.4 Gbp (2,392,715,236 bp)
その理由を Ensembl の help desk に聞いたところ,以下のような返事がきました.概して,Human や Mouse では,染色体 19 番 の LRC/KIR や,染色体数 6 番の MHC 領域などには.他の種類の配列が登録されており,これらがゲノムデータを大きくしている,とのことです.
The genomes of human, mouse and zebrafish are maintained by the Genome Reference Consortium and apart from the primary assembly, they also include alternative sequences (namely patches and haplotypes).
As the primary assembly represents a single version of the genome, known haplotypes that cannot be narrowed down to one sequence are provided as alternative sequences. The current human assembly includes 261 alt loci scaffolds, mainly in the LRC/KIR complex on chromosome 19 (35 alternate sequence representations) and the MHC region on chromosome 6 (7 alternate sequence representations).
You can learn more about patches and haplotypes by waching our video: https://www.youtube.com/watch?v=sPE9j_Hw9HU
Those alternative sequences significantly increase the mouse and human top-level fasta files. If you do not wish to include them in your analyses, you can find primary_assembly.fa files on our ftp site, which do not contain alternative sequences.
このため,通常の local BLAST search では,.toplevel.fa ではなく、.primary_assembly.fa ファイルを使う方が好ましいようです.fasta file のサイズ,BLAST search の速度が大きく異なります.なお,.primary_aseembly.fa ファイルは,Human, Mouse, Zebrafish にしかありませんでした (2019 年 10 月). |
| |
データの
サイズ(GB) |
BLAST 解析
の速度(sec) |
| Homo_sapiens.GRCh38.dna_rm.primary_assembly.fa |
3.2 |
9.3 |
| Homo_sapiens.GRCh38.dna_rm.toplevel.fa |
57.9 |
48.3 |
|
| (2019 年 10 月) |
|
ブラウザを用いたダウンロード
ゲノムデータのダウンロードはこちらです.私は毎回ゲストとして入っています.
ftp://ftp.ensembl.org/pub/current_fasta/
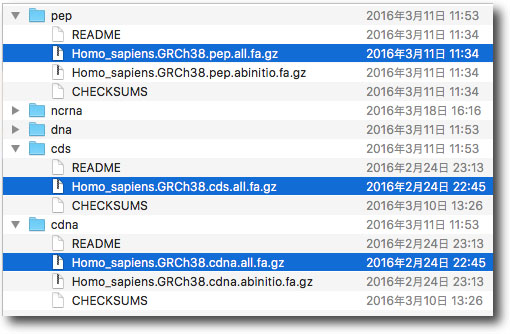
cDNA 配列だけを得ることもできます.
例えば,homo_sapiens では,
cdna/Homo_sapiens.GRCh37.57.cdna.all.fa.gz
の DNA 配列を翻訳すると
pep/Homo_sapiens.GRCh37.57.pep.all.fa
にあるアミノ酸配列が得られます.系統解析を行う場合は,アミノ酸のアライメントをまず作成し,これに従って塩基配列をアライメントします.PAL2NAL を用いることで,塩基配列のアライメントが行えます.PAL2NAL は,開始コドンより前にある余分な塩基配列 (UTRs: Untranslated Regions) を削除して,塩基配列のアライメントを作成します.こちらをご覧下さい.
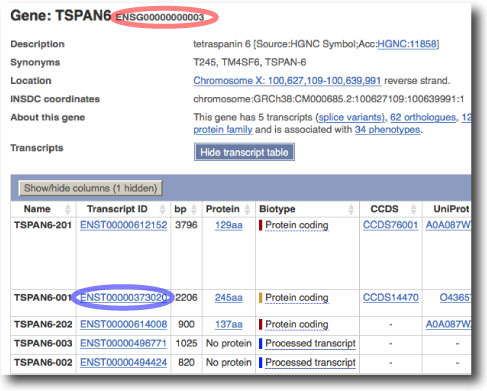
なお,Gene-Protein-Transcript ID はこちらを参照してください.
*最近は CDS (コーディング配列,UTR が取り除かれた DNA 配列) も配布されています (2016 年 4 月). |
|
コマンドラインを用いたダウンロード
ftp を使ってダウンロードすることも可能です (2013 年 6 月).
[Napoli:RawDataEns72]$ ftp
ftp> open ftp.ensembl.org
Connected to ftpservice1.sanger.ac.uk.
220-ftp.ensembl.org NcFTPd Server (free educational license) ready.
220-Ensembl Project FTP server
...
220-
220
Name (ftp.ensembl.org:Napoli): anonymous [anonymous で入ります]
331 Guest login ok, send your complete e-mail address as password.
Password: [メールアドレスを入れるか,単にリターンでも大丈夫です]
230-You are user #92 of 250 simultaneous users allowed.
...
Using binary mode to transfer files.
ftp> cd pub [あとは通常の unix コマンドです]
250 "/pub" is new cwd.
ftp> ls
227 Entering Passive Mode (193,62,203,113,139,113)
150 Data connection accepted from 203.181.243.17:51599; transfer starting.
drwxrwxr-x 2 ftpuser ftpusers 4096 Mar 12 2002 IPI
-rw-rw-r-- 1 ftpuser ftpusers 0 May 1 2002 PLEASE_CHECK_THE...
-rw-rw-r-- 1 ftpuser ftpusers 1877 Oct 22 2007 README.32-46
drwxrwxr-x 5 ftpuser ftpusers 44 May 24 2007 assembly
...
lrwxrwxrwx 1 ftpuser ftpusers 15 Jun 18 12:11 release-72 -> mnt2/release-72
drwxrwxr-x 9 ftpuser ftpusers 109 Dec 4 2002 repository
...
ftp> cd cdna
250 "/pub/mnt2/release-72/fasta/gallus_gallus/cdna" is new cwd.
ftp> ls
227 Entering Passive Mode (193,62,203,113,209,61)
150 Data connection accepted from 203.181.243.17:51607; transfer starting.
-rwxrwxr-x 1 ftpuser ftpusers 128 May 31 04:29 CHECKSUMS
-rwxrwxr-x 1 ftpuser ftpusers 16674832 May 30 23:48 Gallus_gallus.Galgal4.72.cdna.abinitio.fa.gz
-rwxrwxr-x 1 ftpuser ftpusers 13326053 May 30 23:37 Gallus_gallus...
-rwxrwxr-x 1 ftpuser ftpusers 3035 May 30 23:48 README
226 Listing completed.
ftp> get Gallus_gallus.Galgal4.72.cdna.all.fa.gz
local: Gallus_gallus.Galgal4.72.cdna.all.fa.gz remote: Gallus_gallus...
227 Entering Passive Mode (193,62,203,113,146,150)
150 Data connec.....
40% |**********************************
|
Protein coding gene の数を調べる
Ensembl にメールで聞いた方法です.
こちらからそれぞれの種のページに行って,左にある Assembly and Genebuild page を選びます.
例えば Human の場合,こちらに遺伝子の数が書いてあります. Known protein-coding genes と Novel protein-coding genes の値を足した数字が,全タンパク質遺伝子の数です.
Chicken の場合はこちらです.Known protein-coding genes, Projected protein-coding genes, および Novel protein-coding genes の値を足すと,pep.fa ファイルにある ENSGALG のレコード数と一致しました.ただし,うまく数字が合わない種もありました.
実際は database にターミナルなどから入って調べる方法もあるみたいですが,私にはよくわかりませんでした.詳細はこちらを,と紹介されましたが,私はまだ試していません.
Hello Jun,
This is what I get when I query our database:
mysql -u anonymous -h ensembldb.ensembl.org -P 5306
Welcome to the MySQL monitor. Commands end with ; or \g.
Your MySQL connection id is 376882 to server version: 5.1.34-log
Type 'help;' or '\h' for help. Type '\c' to clear the buffer.
mysql> use homo_sapiens_core_63_37;
Reading table information for completion of table and column names
You can turn off this feature to get a quicker startup with -A
Database changed
mysql> SELECT COUNT(*) FROM gene WHERE biotype LIKE 'protein_coding';
+----------+
| COUNT(*) |
+----------+
| 21494 |
+----------+
1 row in set (0.11 sec)
This equals 20599 + 895, as mentioned on http://www.ensembl.org/Homo_sapiens/Info/StatsTable?db=core. So the numbers on this page look allright to me.
Hope this helps.
|
Ensembl Plants の ftp 接続
Ensembl vertebrates では問題になりませんが、Ensembl plants は、ブラウザから ftp 接続が難しいです (Mac の場合)。以下のように、個別の種のページから入れました。そうすると、rsync でダウンロードするアドレスがわかります (2021 年 7 月)。 |
 |
| |
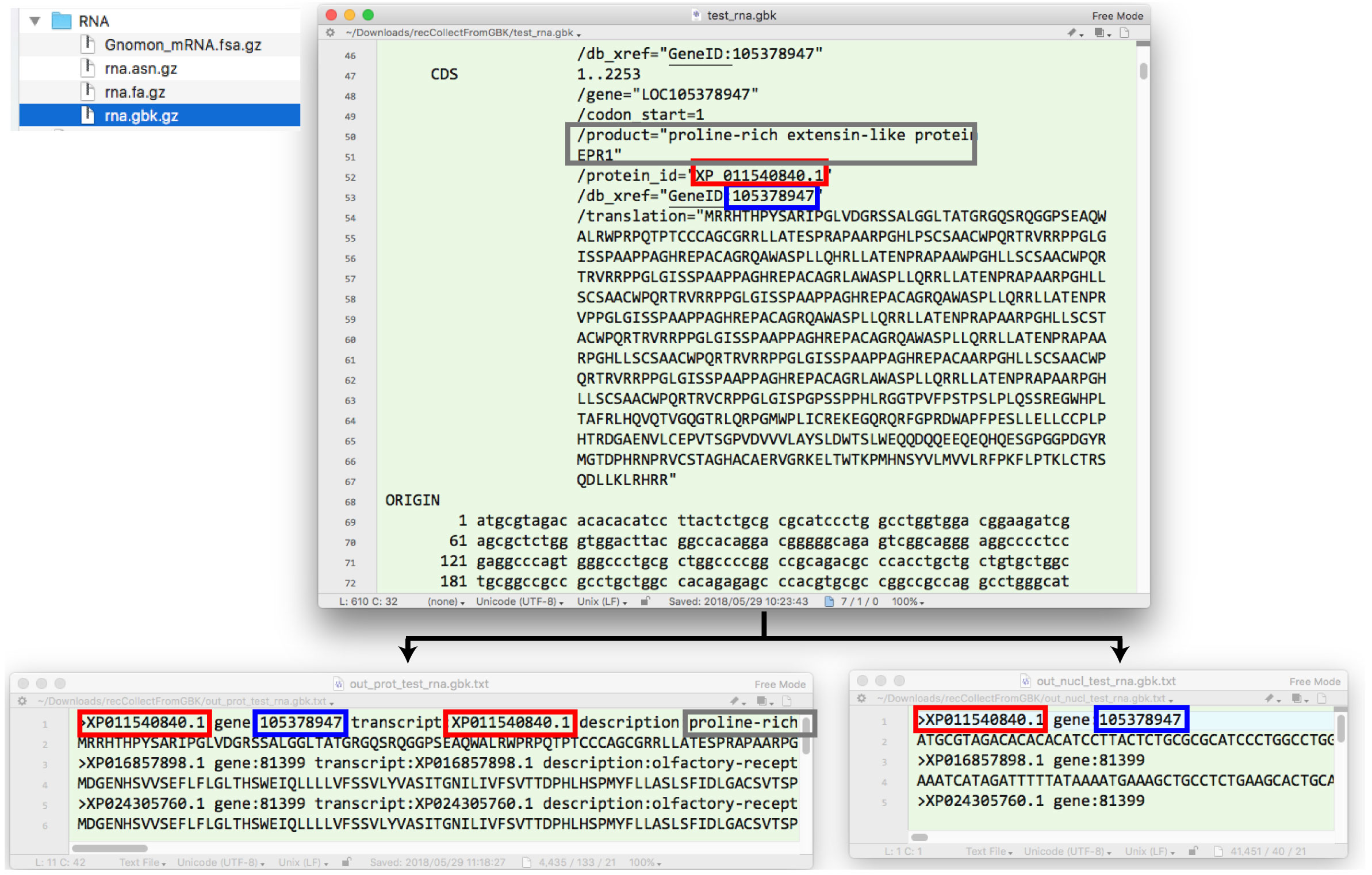
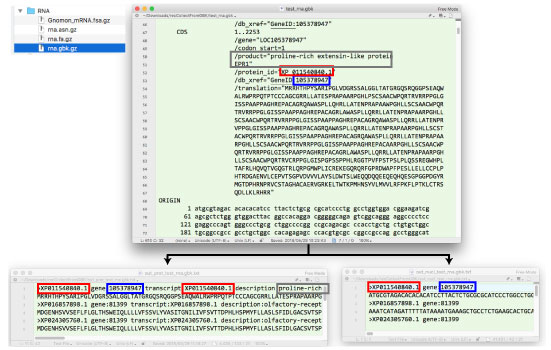
| GenBank 形式から protein と rna レコードを抜き出す |
|
recCollectFromGBK.tar.gz.
(2021/2/1)
NCBI の Human データから例題を作って解析しました.RefSeq データのダウンロードは「RefSeq からデータを集める」を参照してください.(2018 年 9 月). |
|
|
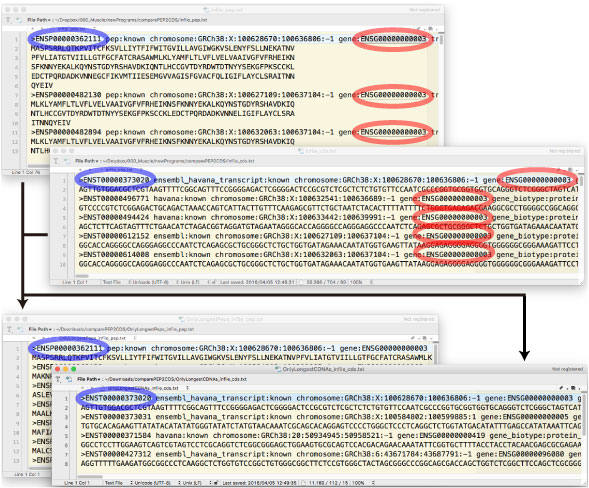
comparePEP2CDS_new.tar.gz (2024/3/3)
comparePEP2CDS.tar.gz (古いバージョン。遅いです。2021/2/1).
Chicken E104 データ (28444 records, 16878 genes) を、最新版は 4 分、古いバージョンは 19 分かかりました。
gene ID (e.g., ENSG0000001) から最長のトランスクリプトームを抜き出します.その後,PEP と DNA レコード (cDNA あるいは CDS レコード) を pal2nal (v14) で比較し,一致しなければ翻訳PEP レコードを補正します.補正は 3 点を行います.
- U を * に変換.
- 配列最初が X (がいくつか) あるなら,これらを削除.
- あるいは末尾のアミノ酸残基を削除.
それでも補正されない場合は,050_out.txt に出るエラーメッセージに従って,一つ一つ検証してください.
Human のデータは 10 分ぐらいかかりますが,魚類などのデータでは数分で解析が終わります.Fasta file の name line は,geneID や protein ID が Ensembl のスタイルで書かれている必要があります.
解析が無事に終了すると,pal2nal の出力として,050_out.txt に以下のようなアウトプットが出ます.エラーメッセージが表示されません.
CLUSTAL W multiple sequence alignment
ENSP00000362111 ATGGCGTCCCCGTCTCGGAGACTGCAGACTAA
ENSP00000362122 ATGGCAAAGAATCCTCCAGAGAATTGTGAAGA
ENSP00000360640 GCCTCCTTGGAAGTCAGTCGTAGTCCTCGCAG
Fasta file の name line は,geneID や protein ID が Ensembl のスタイルで書かれている必要があります.
解析手順:
perl control.pl
アウトファイル:
000_OnlyLongestCDNAs_infile_cds.txt
000_OnlyLongestPeps_infile_pep.txt
|
|
| Web で確認できます. |
|
|
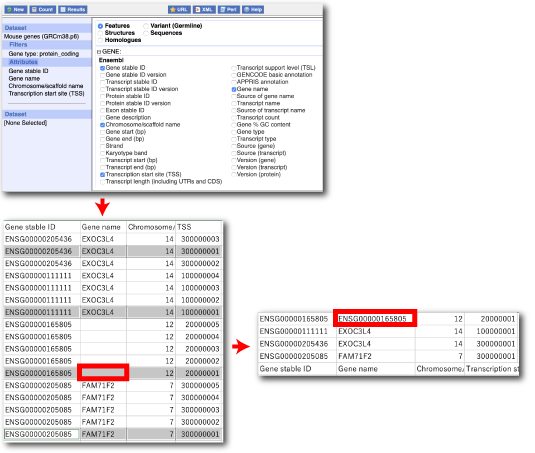 |
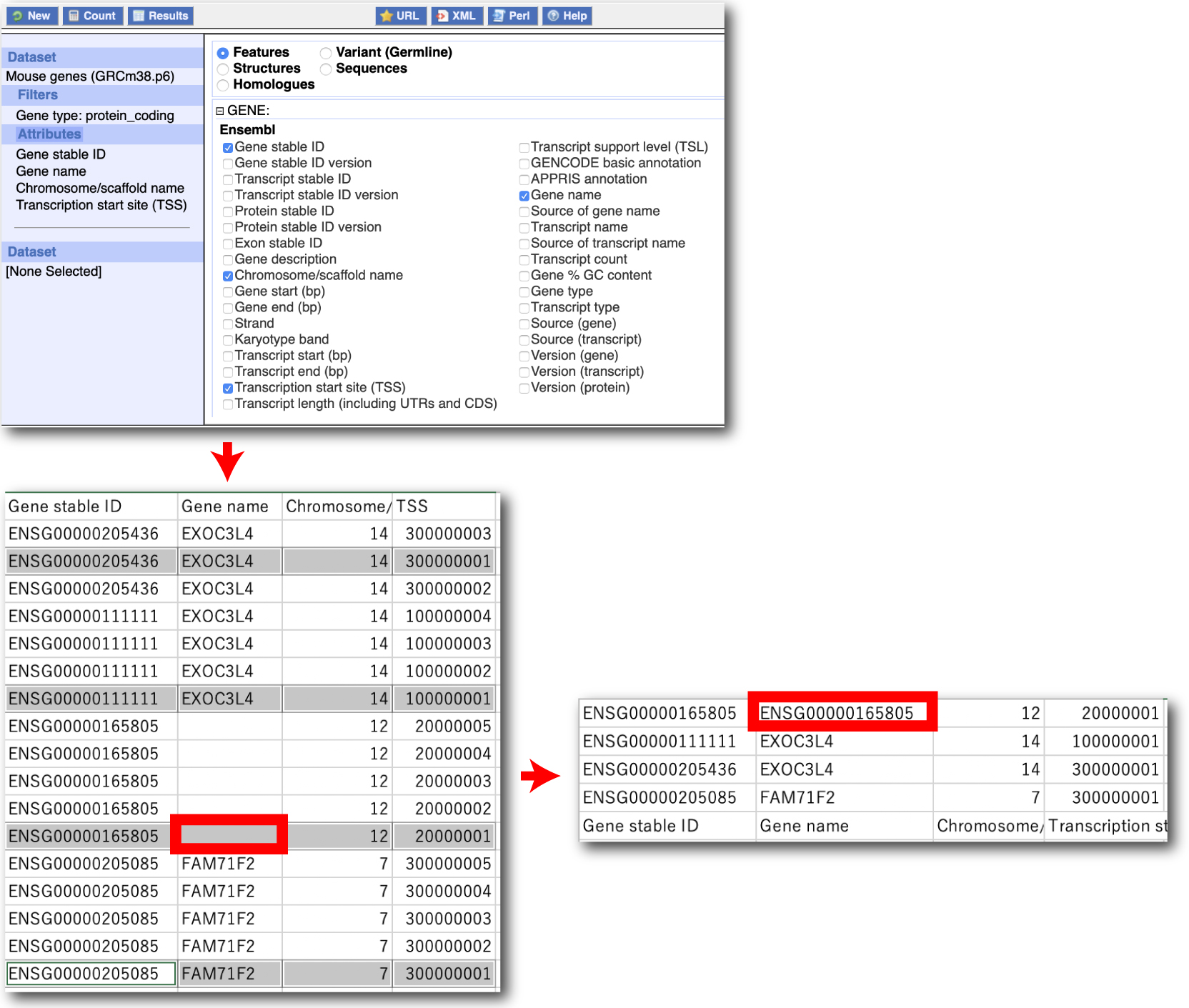
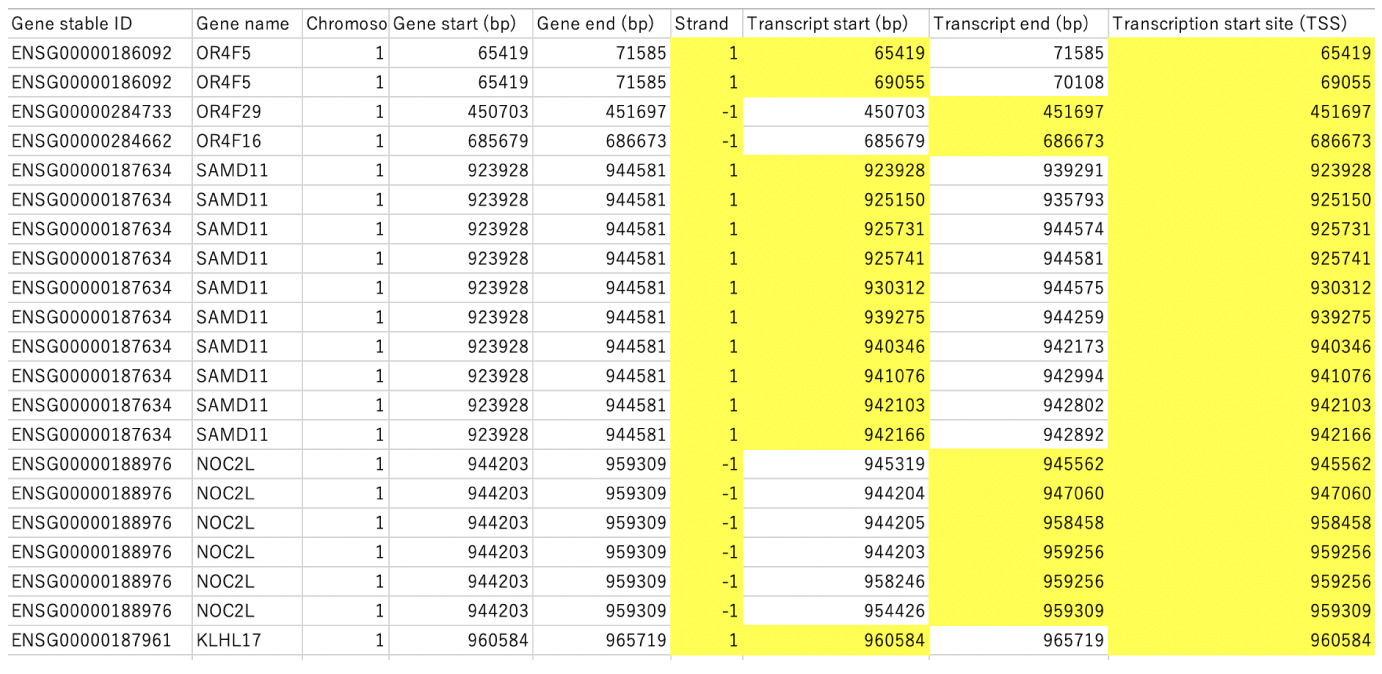
selectTSS.tar.gz
BioMart では,TSS は Strand に従い, Transcript start (1) かあるいは Transcript end (-1) が選ばれています (以下). |
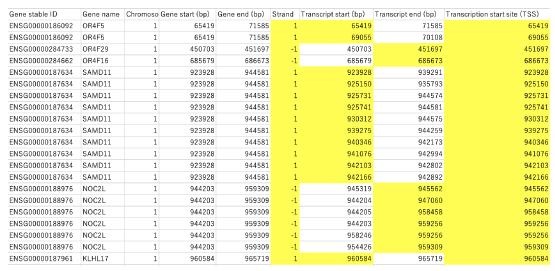 |
2019 年 10 月
|
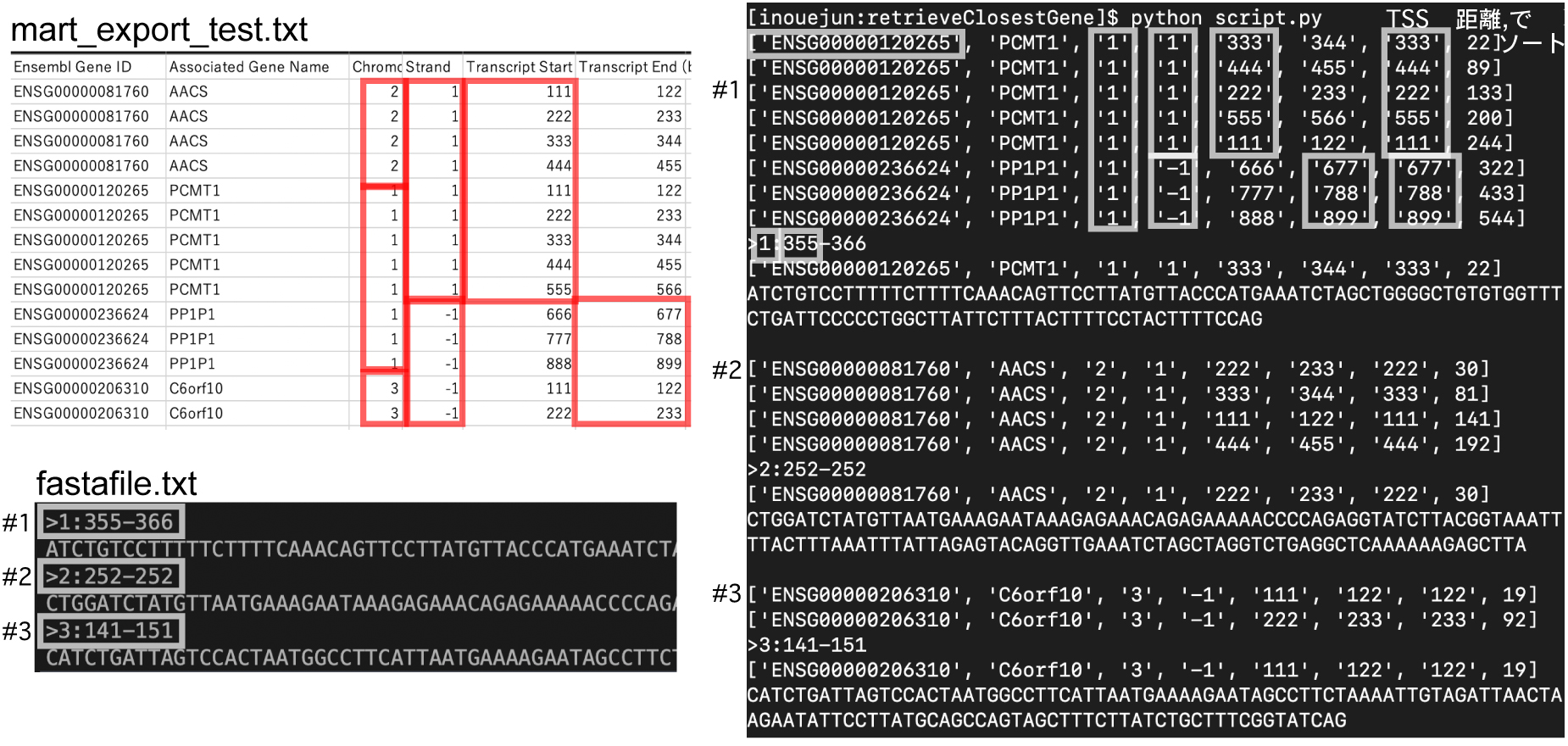
| BioMart: ある座標にもっとも近い遺伝子のレコードを得る-TSSあり |
|
 |
retrieveClosestGene_TSS.tar.gz
2019 年 9 月
|
| BioMart: ある座標にもっとも近い遺伝子のレコードを得る-TSSなし |
|
 |
retrieveClosestGene.tar.gz
2019 年 9 月
|
|
| Exon 配列を取得する |
GTF ファイルにすべての情報が集約されています.GTF ファイルについては,こちらを参照してください.その例として,ダウンロードした DNA と GTF ファスタファイルを使って,Protein ID (or Gene ID) に基づいてエクソン配列を得るスクリプトを紹介します.
[i2:zebraGTF]$ perl gtfGrep.pl ENSDARP00000053510
>ENSDARP00000053510_Exons0
ATGGAGAGCGCTCTGTCGGCCAGAGACCGGG.....
>ENSDARP00000053510_Exons1
ACATATATTGGGTCGGTGCTGGTGTCGGTGA.....
.....
zebraGTF.tar.gz [33.2MB]
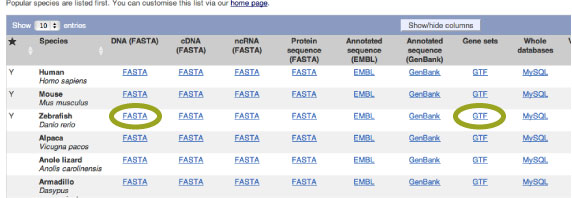
以下 (FTP Download) から必要なファスタファイルをダウンロードしてください [2013 年 5 月].
|
|
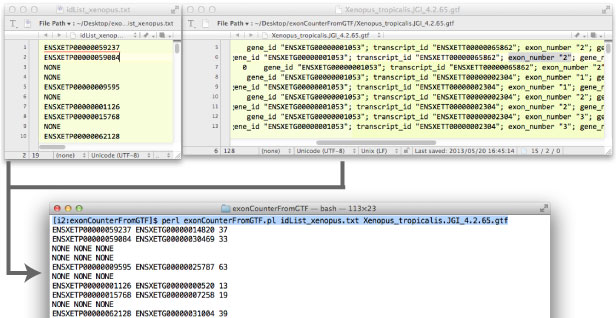
遺伝子ごとのエクソン数を調べる |
Protein ID のリストに従って,その遺伝子すべての Exon 数を数えます.Protein ID に対応した Exon 数ではありません [2016 年 7 月].
exonCounterFromGTF.tar.gz
BioMart を使う方法はこちらをご覧ください.
|
|
|
| TSS 情報を取り出す |
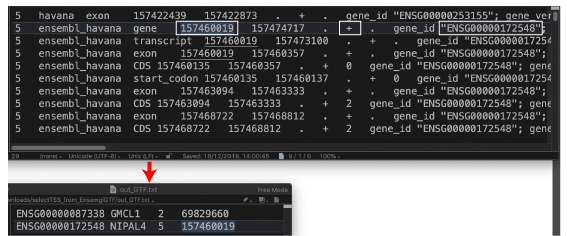 |
GTF ファイルから TSS 情報を取り出すスクリプトです。
selectTSS_from_EnsemglGTF.tar.gz
Ensembl が提供する GTF ファイルはこちらからダウンロードできます。
ftp://ftp.ensembl.org/pub/release-98/gtf/homo_sapiens/
(2020 年 1 月)
|
|
BioMart を用いない Ensembl ゲノムブラウザのを使い方を一部だけ紹介します.
|
核タンパク質コーディング遺伝子領域のアライメント
Ensembl のウェブサイトを使って,アライメントを作成できます.統合テレビ「Ensembl tips 〜塩基配列のアラインメントを作成する〜2009」に解説がありました.
ここでは,複数種からなる Nestin 遺伝子のゲノムアライメント作成方法が紹介されています.5' や 3' 上流域をアライメントに含めるやり方も紹介されています.
ただし,ある程度使えるのはヒトとその周辺の哺乳類まで,という感じです.複数種のアライメントは,哺乳類を中心に作られた既存のアライメントしかありません.ペアワイズであれば他の種でもできるようですが,例題であげられた Nestin 遺伝子であっても,Human vs Zebrafish ではおかしなペアワイズアライメントが示されました [2012 年 12 月].
ある遺伝子周辺のシンテニーを種間で比較する
統合テレビ「Ensembl tips 配列を比較する 2011」に詳しく紹介されています.
ここでは ADIPOQ 遺伝子とその周辺のゲノム構造をヒトとマウス,イヌ,と比較する方法が紹介されています.配列のアライメントは哺乳類を中心にあらかじめ作られたものしか表示されません.あとはペアワイズです [2012 年 12 月].
|
|
オーソログや曖昧な DNA 配列 (R や Y) については,こちらにまとめられています.
|
|
|
Ensembl と NCBI のデータベースは完全には共有されていない
Ensembl で公開されている配列が NCBI の Blast search で引っかかって来ないことがよくあります.Ensembl の担当者にきいたところ,以下のような返事が帰ってきました.
The Ensembl gene set is based on mRNA and protein sequences from NCBI RefSeq (only the known protein, NP, and known mRNAs, NM) along with UniProt and EMBL-Bank. For a more detailed description, see this page:
http://www.ensembl.org/info/docs/genebuild/genome_annotation.html
Thus, the Ensembl geneset contains some NCBI RefSeq records (those that can be aligned to the genome). However, not all RefSeq records. And RefSeq does not have all Ensembl transcripts.
Variation について
遺伝子によっては Variant がある場合があります.例えば,PSMB8 遺伝子は Human では 1 コピーしかなく 6 番染色体にコードされているはずですが,Location が HSCHR6 になっている Variation が示されます.こちらです.
これについてヘルプで救い問い合わせたところ,Biomart から Variation table を得る必要がある,という解答を得ました.
Ensembl Blog にも関連の記事がありました (2014 年 11 月).
Depending on the number of genes you're looking at, you can identify the variants in a few different ways. If you're only looking at one or two, you may find it easier to search for the genes within the browser, then open the variation tables. Another way to access variants in genes may be through BioMart. This allows you to download tables of data. There's a video tutorial on using BioMart here. You will need the human variation database. For genes on different haplotypes, you will need to search by all the associated Ensembl IDs.
Alternatively, you can extract the data using our Perl API, particularly if you are looking at a large number of genes. Use these links to install the API, and find tutorials on its use:
http://www.ensembl.org/info/docs/api/api_installation.html
http://www.ensembl.org/info/docs/api/core/core_tutorial.html
http://www.ensembl.org/info/docs/api/variation/variation_tutorial.html
There is a list of the different haplotype names at the MHC region at:
http://www.ensembl.org/Help/Faq?id=291
|
pep all ファイルは転写産物の配列のみ
ダウンロードできる全タンパク質遺伝子の配列は,以下の二種類があります (homo_sapiens/ pep ディレクトリ).
ftp://ftp.ensembl.org/pub/current_fasta/
Homo_sapiens.GRCh38.pep.all.fa
Homo_sapiens.GRCh38.pep.abinitio.fa
all.fa ファイルには,転写産物 (mRNA とタンパク質配列) から得られる配列のみが含まれているため,発現量がごく微量であったり発現されないタンパク質遺伝子の配列は含まれていません.一方 pep ab initio ファイルにはゲノム配列からタンパク質遺伝子の配列を推定しているようですが,README を読む限り,他の情報と照らし合わせて確認はしていないようです.フグ (ver76) で両者のレコード数を比較すると,pep all が 23118 とおよそ全タンパク質遺伝子数 20,000 であるのに対し,pep ab initio は 48390 となっているので未整理な情報がかなり含まれているようです.
標記の件に付いては,Ensembl に問い合わせて確認しました.
###### メール 1
See an example here:
ftp://ftp.ensembl.org/pub/release-75/fasta/homo_sapiens/pep/
a) pep all: resulting from Ensembl known or novel gene models which are based on transcriptome and proteome data
b) pep ab initio: resulting from 'ab initio' gene prediction algorithms solely based on the genomic sequence with no other experimental evidence.
More information on these files can be found in the README
ftp://ftp.ensembl.org/pub/release-75/fasta/homo_sapiens/pep/README
The same applies to cdna all versus cdna ab inition:
ftp://ftp.ensembl.org/pub/release-75/fasta/homo_sapiens/cdna/README
cdna.all.fa and pep.all.fa should not contain ab initio predictions.
Kind regards,
###### メール 2
The “pep all" file includes ONLY the peptide sequences derived from transcriptome (mRNAs) and proteome data.
It does NOT include sequences predicted from ab initio methods.
There must be proteins that cant be annotated to their low level of expression.
“pep all” might be a misnomer and we are aware of that. I will bring this to my colleagues and see if this name can be changed.
Regards,
######
(2014 年 8 月)
|
古い ID について
(以下のように書きましたが,あまり有効な手段ではありません.最近は古い配列をクエリとして最新のデータベースを Blast 検索しています.それ以外方法はないように思えます [2011 年 11 月].)
古い ID から最新の ID を得る
BioMart を使います.
1. 古い ID が得られた version の BioMart から Entrez の ID を得ます.
例えば BioMart v36 に入ります.ポップアップタグを「Ensembl38」「Homo sapiens genes (NCBI36)」にします.
Next をおして,GENE -> ID list limit をチェックし,以下の例題をボックスにペーストして,ポップアップタグを Ensembl Peptide ID(s) にします (ID をリストアップしたファイルを選ぶやり方は,うまくいきませんでした).Next を押します.
ENSP00000277994
ENSP00000252073
ENSP00000354686
ENSP00000354852
FILTER 画面になります.External Referece: から EntrezGene ID をチェックし,Select the output format: の Text, tab separated,File compression: の gzip (.gz) をチェックして export を押します.結果がテキストファイルとしてダウンロードされます.
2.
これを最新の BioMart で新しい ID に変換します.
以下の EntrezGeneID が得られているはずです.
22834
63916
4288
6263
ヒトデータ指定した最新の BioMart を開きます.こちらです.Filter -> GENE から ID list limit をチェックします.ボックスに上記の EntrezGeneID をペースとしてポップアップタグを EntrezGene ID(s) にします.Attributes -> EXTERNAL: -> External References から EntrezGene ID を選びます.
あとはいつも通り,Count を押して ID 数を確認し,Results を押して,Unique results only をチェックし,Go を押すことで,結果をテキストファイルとしてダウンロードできます.
History を調べる
ID の変更履歴を調べることができます.ただし,通常の作業ではあまり必要ないと思います.
こちらから名前のかわった ID (新旧) について調べることができます.
Manage your data というタブをこちらから押してください.
その後,ID History Converter を押します.
ただこの方法だと,一度に 30 件までしか調べられないようです.
それと,実際に website で一つずつ調べた結果に比べて,得られる結果の精度と使いやすさが落ちるようです.
|
| Perl script |
種ごとのデータベースから最長のトランスクリプトだけを集める
こちらから全タンパク質コーディング遺伝子をダウンロードする場合,まずアミノ酸配列のファイル (pep.all.fa.pin) を処理することになります.pep.all.fa.pin ファイルでは (対応する cDNA データベースも同じですが),一つの遺伝子について複数のトランスクリプトが書かれていることが多いです.このような場合に,一つの遺伝子から一つの配列だけを選ぶために,最長のトランスクリプトを抜き出すスクリプトを作りました.以下のようなスクリーンアウトが出て,アウトファイルが作成されます.
[inouejun:longPepPicker_fol]$ perl longPepPicker.pl DB.pep.faFile name: DB.pep.fa
number of records: 11
number of ensg ids: 4
number of ensp ids: 11
number of enst ids: 11
ちなみに,Human の .pep.fa ファイル (Ensembl v61) は,10 分ほどかかりました.
File name: Homo_sapiens.GRCh37.61.pep.all.fa
number of records: 86934
number of ensg ids: 21818
number of ensp ids: 86934
number of enst ids: 86934
longPepPicker_fol.tar.gz [2014 年 1 月 改訂].
|
pep レコードに対応した cdna レコードを集める
上記のスクリプトで,最長のトランスクリプトだけからなる pep データベースを作った場合などに,対応する cdna レコードを cdna データベースから拾ってくるスクリプトです.
cDNApickerFromPep_fol.tar.gz [2014 年 1 月 改訂].
|
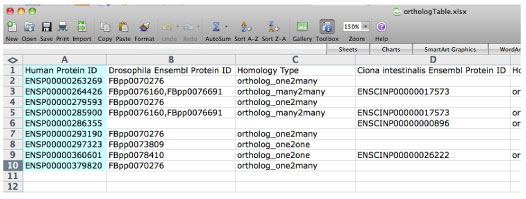
DB から ID の配列を取ってくる
BioMart から得た Human の orthologue information (下図) に従って,それぞれの種の DB から対応する ID の配列を取ってくるスクリプトです.
BioMart からダウンロードした Table を Excel で保存して,Human の ID に対応した他の種の ID を同じ行に列挙してください.Human の ID でアウトファイルが遺伝子グループごとに作成されます.一行目は Human でなくても動くはずです.
詳しくは例題である orthSeqPicker_fol.tar.gz を参照してください [2011 年 3 月]. |
|
pep レコードに対応する cDNA レコードを集める
.pep.all.fa の最後にある ENST ID をキーワードにして cDNA データベースを検索します.
トランスクリプトームデータはアミノ酸配列 (.pep.all.fa)と,これに対応する cDNA 配列 (.cdna.all.fa) のデータベースがあります.それぞれ name line は以下のようになっています.
.pep.all.fa
>ENSP00000374981 pep:known chromosome:GRCh37:14:106053226:106054732:-1 gene:ENSG00000211890 transcript:ENST00000390539
.cdna.all.fa
>ENST00000390539 cdna:known chromosome:GRCh37:14:106053226:106054732:-1 gene:ENSG00000211890
ensDNAseqRet_fol.tar.gz
ID をたよりに pep ファイルからレコードを集める
ENSP number はわかるけれど,対応する ENT number がわからない場合,もう一度 pep ファイルからオリジナルの name line (ENSP と ENST number の両方が書いてある)のレコードを集め直します.そうすると,上記のスクリプトで,ENSP レコードに対応した ENST レコードを集めることができます.
例えば,
ID ファイル.
ENSP00000374981
ENSCINP00000001850
ENSDARP00000101617
ensIDseqRet_fol.tar.gz
|
Ensembl: Collecting Orthologs
系統解析に利用可能な相同遺伝子グループを集めるスクリプト集を作りました.こちらです.ただ,こちらは Ensembl が提供する大まかなオーソログ情報に基づいたものであることに注意してください.BioMart と fasta 形式でダウンロードできる種別データベースを用いています [2012 年 12 月].
|
|
NCBI が提供している BLAST+ で,Ensembl が配布している種ごとのゲノムデータを解析することが可能です.Human を例に説明します.
Human の ftp site (ftp://ftp.ensembl.org/pub/release-96/fasta/homo_sapiens/dna/) から,Homo_sapiens_GRCh38.dna.toplevel.fa.gz をダウンロードし解答します.これを NCBI BLAST+ のページから得た makeblastdb, blastn, blastdbcmd などで解析します.BLAST+ の利用方法は,こちらを参照してください.
# Ensembl のゲノムデータは,データベースの作成は非常に時間がかかります.さらに,染色体ごとにファイルが作成されるので,たくさんのファイルになります.NCBI refseq はファイル数も少ないので,やりやすいです.
./makeblastdb -in Homo_sapiens.GRCh38.dna.toplevel.fa -dbtype nucl -parse_seqids
# blastn 解析です.
./blastn -query 000query.txt -db Homo_sapiens.GRCh38.dna.toplevel.fa -num_alignments 10 -evalue 1e-12 -out out_nuclREC_EMBL.txt
# ヒットした配列を得ます. 染色体ごとに別のファイルが作成されますが,複数のファイルを指定することなく,以下のコマンドで問題ないようです.-entry や -range は out_nuclREC_EMBL.txt ファイルに保存される値です.
./blastdbcmd -entry 22 -db Homo_sapiens.GRCh38.dna.toplevel.fa -dbtype nucl -strand plus -range 33168984-33169086 -out out_nuclSEC_EMBL.txt
上の解析で使ったコマンドとクエリ配列はこちらです (blastP_EnsDB.tar.gz).Ensembl から XXX.toplevel.fa ファイルをダウンロードして使ってください (2019 年 7 月).
|
|
Ensembl Tutorials and Worked Examples
Ensembl ミニコース
Ensembl in Singapore
2014 年 1 月の段階で,Hinxton (本家) のデータより最新のゾウギンザメとフグのデータが公開されていました.
|
|