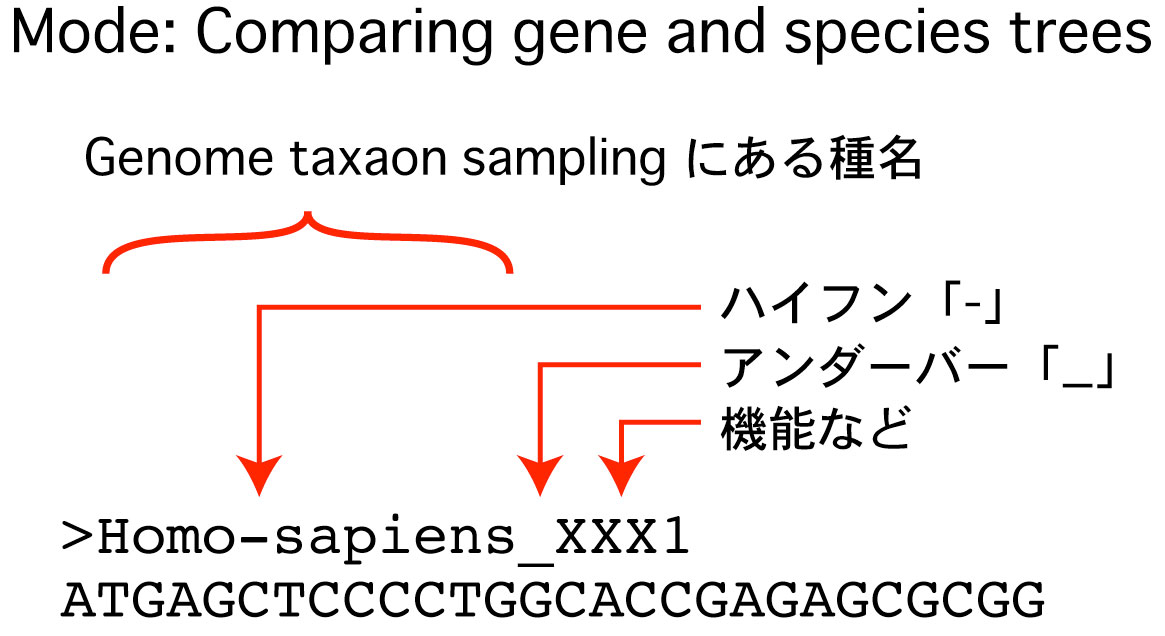
|
|||||||
2024 年 2 月 2 日 改訂 |
|||||||
ORTHOSCOPE は遺伝子系統樹を推定して、異なる種の間で同じ機能の遺伝子 (遺伝子座) を判定するウェブツールです。このため、同じ機能を持った遺伝子が、他の種に存在するか、あるいは何個あるかカウントできます。 ユーザーは、解析の対象とする種を、左右相称動物約 500 種あるいは植物約 50 種から選べます。解析には、DNA 配列 (あるいはアミノ酸配列) をクエリ配列として用います。 まずは、統合テレビによる解説や以下のスライドご覧ください。 |
|||||||
 |
|||||||
|
|||||||
Execute 後の ORTHOSCOPE 自動解析の流れ (1) クエリ配列に類似した配列を、複数種のゲノムデータから収集。 (2) 収集した配列に基づいて、遺伝子系統樹を推定。 (3) 種系統樹と比較して、オーソグループを判定
|
|||||||
|
|||||||
|
|||||||
|
|||||||
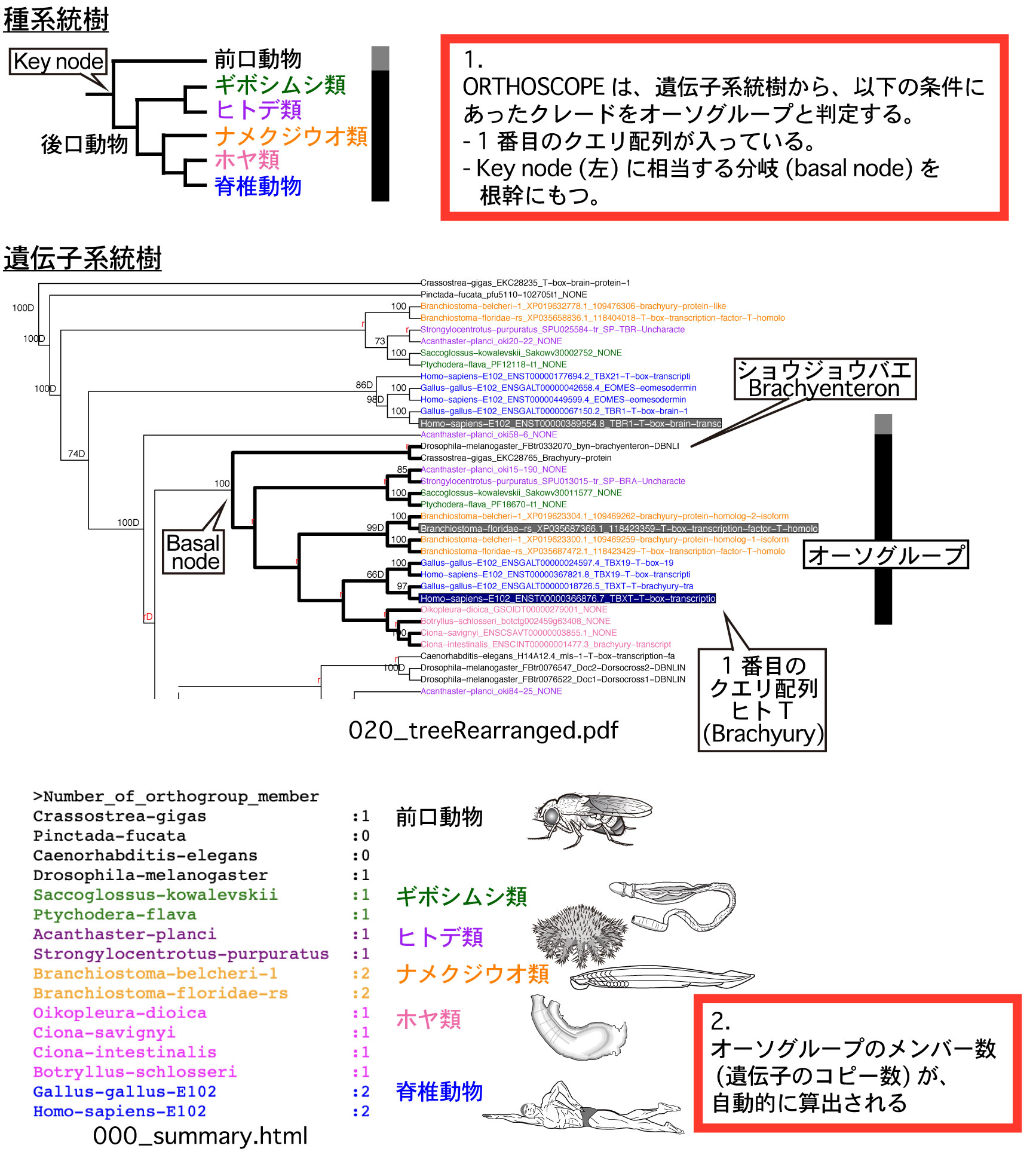
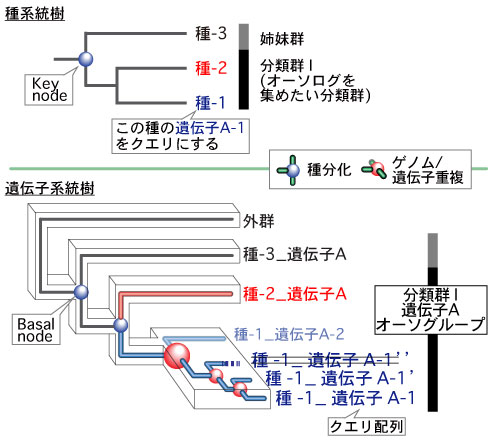
ORTHOSCOPE の機能推定は、オーソグループ (orthogroup) に基づくものです。 |
|||||||
|
|||||||
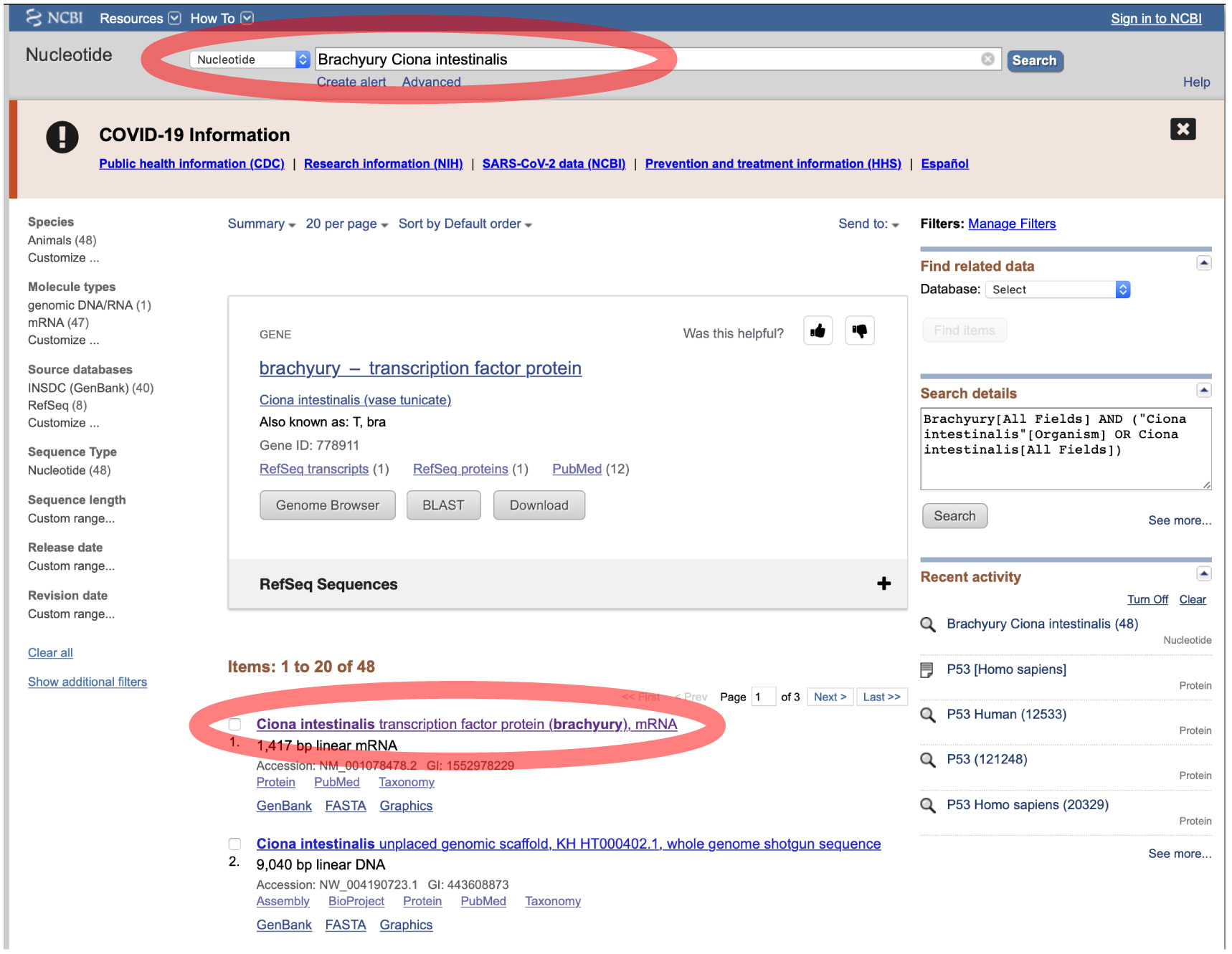
クエリとなる遺伝子配列は、NCBI や Ensembl から得られます。以下は、NCBI から配列を得ています。「Brachyury Ciona intestinalis」をキーワードとして検索し、検索から得られた配列をコピーして ORTHOSCOPE 解析に使ってください。ORTHOSCOPE 解析はコーディング配列 (CDS) の解析を想定しています。 |
|||||||
 |
|||||||
 |
|||||||
|
|
|||||||
|
|||||||
10 人を超えるような授業で同時に解析を行う場合は、サーバーの負荷を減らすために、ミラーサイトを使い分けてください: 1〜6 月生まれの人:viento https://orthoscope.jp/orthoscope/Deuterostomia.html 7〜12 月生まれの人:osaka http://133.167.86.72/orthoscope/Deuterostomia.html
ガン抑制遺伝子のコピー数を数える 1. ゾウのコピー数 2. ハダカデバネズミのコピー数 哺乳類なので、Focal group を Mammalia で解析してください。 答え:result3658.zip、解説。
名前が類似した 3 遺伝子のうち、どの 2 つが進化的に近いか調べる The GLIS family transcription factors, GLIS1 and GLIS3, potentiate generation of induced pluripotent stem cells (iPSCs), although another GLIS family member, GLIS2, suppresses cell reprograming. Using ORTHOSCOPE, Yasuoka et al (2019) showed that GLIS1 and GLIS3 originated during vertebrate whole genome duplication, whereas GLIS2 is a sister group to GLIS1/3. Let's Make sure GLIS gene relationships using ORTHOSOCPE. Answer: result3661.zip. Comments.
Fad2 遺伝子は淡水魚でコピー数が多い? Ishikawa et al. (2019) は、淡水に侵入するイトヨでは、海域に生息するイトヨよりも、DHA 合成に関わる Fads2 遺伝子のコピー数が多いことを見出した。それでは、真骨類では一般的に、海水魚よりも淡水魚の方が Fads2 遺伝子のコピー数が多いと言えるだろうか? ヒント: Instruction の「Example Data: Ishikawa et al (2019)」を参照。
ホヤ CesA 遺伝子は本当にバクテリアから水平伝搬してきた? ホヤは動物界で唯一、セルロースを合成できる CesA 遺伝子を持っている (笹倉研究室)。ホヤは成体になると、セルロースでできた殻で体を覆って岩などにくっついてしまい、植物のような生活を送る。Nakashima et al. (2004) は、ホヤがCesA 遺伝子をバクテリアから水平伝搬によって得たと推定した。それが本当なら、少なくともホヤ類以外の動物は、CesA 遺伝子を持っていないはずである。ORTHOSCOPE 解析で、そのことを確かめてみましょう。 ヒント: Instruction の「Example Data: Inoue et al (2019)」を参照。
|
|||||||
|
|||||||
DeuterostomeBra_2ndAnalysis.zip この解析パイプラインは、ダウンロードして利用します。ORTHOSCOPE 解析の結果を元に、オーソグループのメンバーに絞った系統樹を推定できます。スクリプトは Mac 解析用に Python3 で作成しました。Windows ユーザーはスクリプトを若干改訂する必要があるかもしれません。 |
|||||||
必要なプログラムのインストール 系統樹を推定するには、いくつかの解析プログラムをインストールして、パスを設定する必要があります。
パスにアドレスを追加します。例えば、
trimAl v1.2 (Official release):
|
|||||||
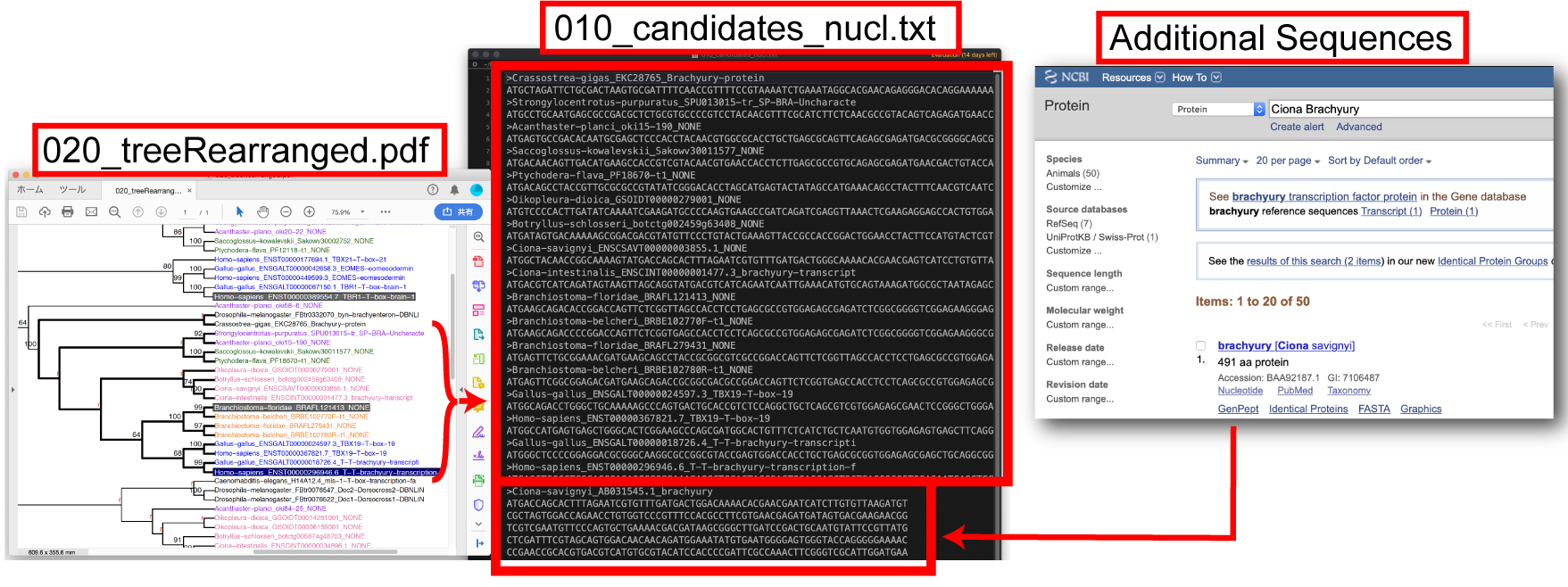
系統樹の推定 1. 適切なアウトグループとオーソグループメンバーを 010_candidates_nucl.txt file に保存します。アウトグループとなる配列は、アライメントの一番最初に置いてください。NCBI などから得た配列 (additional sequence) をここで加えることができます。 |
|||||||
 |
|||||||
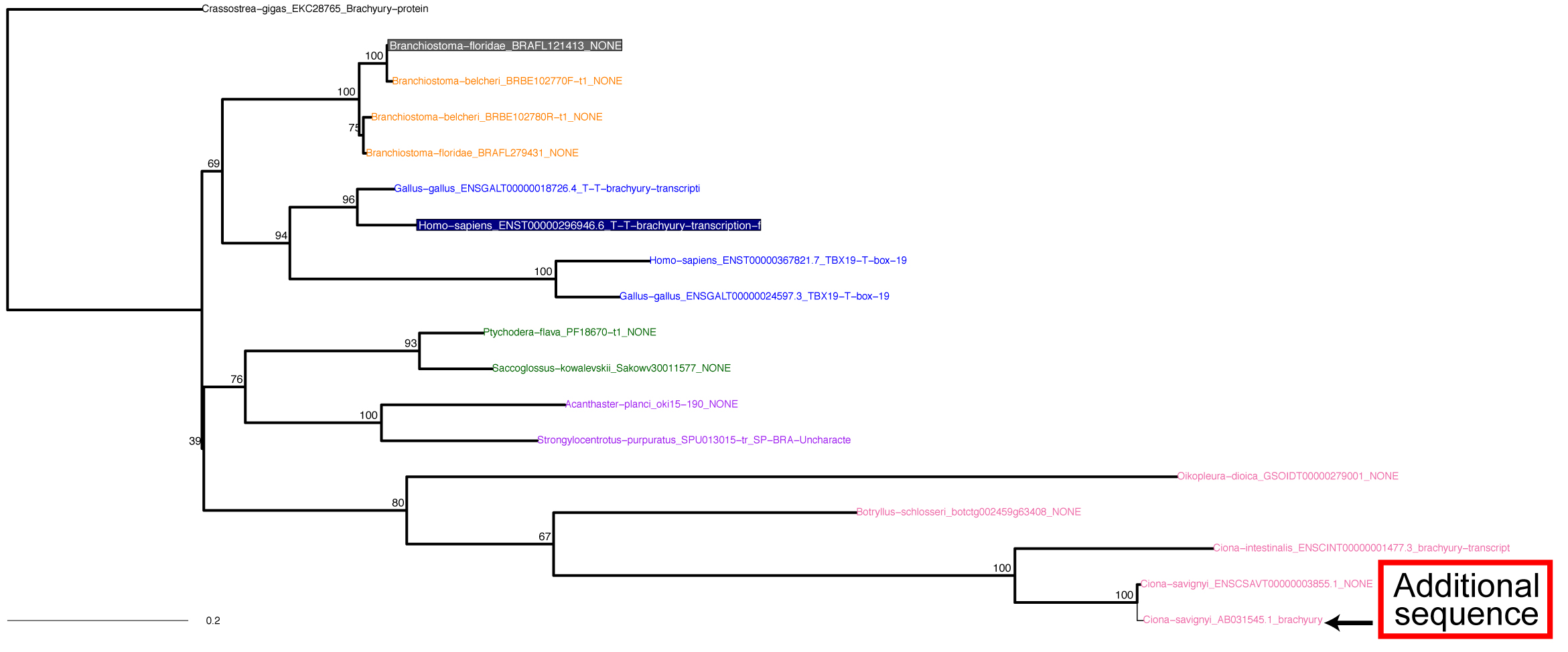
2. 100_2ndTree.tar.gz ファイルを解凍してください。
5. ML tree は 200_RAxMLtree_Exc3rd.pdf に保存されます. |
|||||||
 |
|||||||
|
|||||||
|
|||||||
|