|
|
|
|
Notung は species tree (種系統樹) と gene tree (遺伝子系統樹:一つの種から複数のホモログを含む) を比較して,遺伝子重複と欠失が生じたノードを最節約推定するプログラムです. Notung の最節約原理は,遺伝子重複と遺伝子欠失が滅多に生じない現象であるとするもので,これらができるだけ少なくなるような位置に遺伝子重複/ 欠失を配置したり,gene tree を再編成します.Species tree は文献等を参照して作成しておきます.Gene tree は RAxML などであらかじめ推定しておく必要があります.
単に gene tree と species tree の比較だけを行うのは Reconciliation Mode です.Gene tree においてブートストラップ値などの低いノードを,species tree と比較して補正する機能は Rearrange Mode と言います.
マニュアル (145 ページ) には,使い方が丁寧に書いてあります.適切な例題もあります.Graphical user interface (GUI) version もありますが,ここではマックの terminal を用いた command line interfece (CLI) version を紹介します.
遺伝子の系統樹と種の系統樹を比較する解析は,今後さらに重要さが増していくように思えます.現在でもベイズ法を用いたソフトウェアなどがあるようですが,私が調べた限りでは Notung が最も使いやすく現実的な解析のように思えます [2011 年 11 月].
|
|
- 初めての方は command line option をやる前に,GUI でマニュアル「Appendix E」にある Worked Examples をやってみた方が良いです.
- Command line option で Rearrange の解析を行うと,自動的に reconciliation も行ってくれます.このため Rearrange を行った場合は新たに reconciliation を行う必要はありません.
- Notung は系統樹のテキスト表示に New Hampshier X (NHX) フォーマットを使っています.使っているうちにだいたい意味はわかると思いますが,詳しくは以下を参照してください.
http://home.cc.umanitoba.ca/~psgendb/doc/atv/NHX.pdf
https://sites.google.com/site/cmzmasek/home/software/forester/nhx
|
| Reconciliation Mode (Batch File) |
|
(*私はバッチファイルは使っていません.Perl script と組み合わせて command line を使っています [2011 年 9 月]).
得られた gene tree を species tree と照らし合わせて,遺伝子重複が生じた node を推定します.こちらにどのような解析が行われるか図で表されています.
マニュアル P83 「12.3 Running Notung from a Batch File」に従って,ダウンロードして得られる sampleTrees フォルダーに入っている例題を用いて解析方法を説明します.ここでは,種系統樹と,複数の遺伝子系統樹を比較して,遺伝子重複が生じたノードを推定しています.
解析を走らせる
ターミナルでダウンロード・解凍した Notung フォルダに入ります.その後,
java -jar Notung-2.6.jar -b sampleTrees/batch/batch.run --reconcile --info --usegenedir --speciestag prefix
と入力してください.batch ファイルには,存在しない gene tree などもあって,解析が正常に作動した tree とエラーが出る tree があります.これはエラーを示すためにわざと入っています.
Infiles
batch.run ファイルには,species tree のファイル名,その下に gene tree のファイル名が書かれています.系統樹のファイルは newick 形式で保存します.
結果
b. b という遺伝子系統樹を例として結果を見てみましょう. Notung-2.6.jar をダブルクリックして起動し,File > Open Gene Tree から b.b.reconciled を選んで開きます.Display Options > Display Internal Node Names を選び,node の名前を表示します.これは自動的に名前が付けられるようです.
b.b.reconciled.info というファイルを開きます.左側に Duplication が生じたと考えられる node gが書かれています.実際には L. bound と U. bound で示された node の間で遺伝子重複が生じたことが示されています.
|
| Reconciliation Mode (Command Line) |
|
バッチファイルを用いない command line option の例題を作りました.こちらの方が実用的な気がします
sample_recon.tar.gz
|
|
結果は,NHX フォーマットで保存されます (genetree.tre.reconciled ファイル).
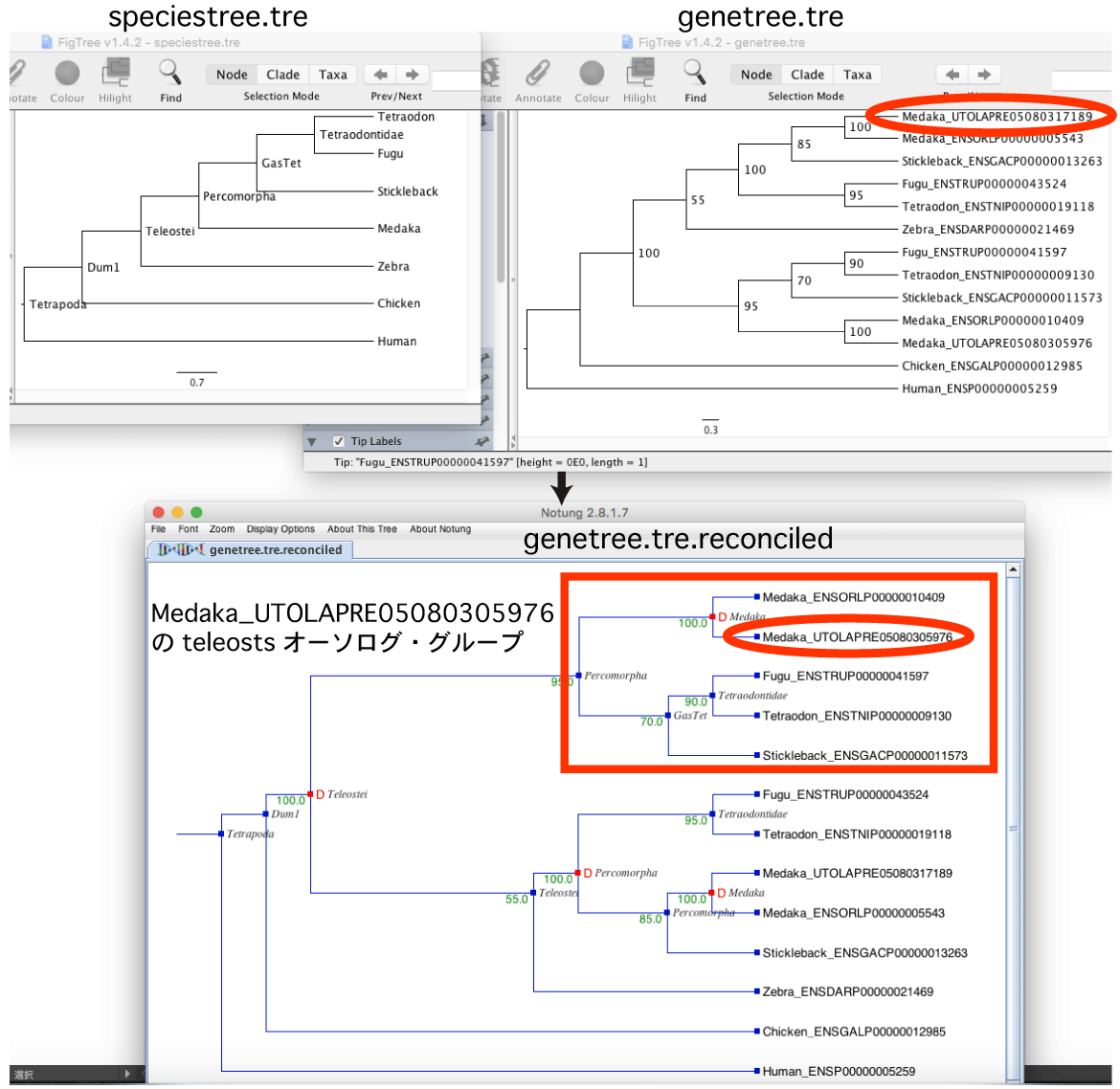
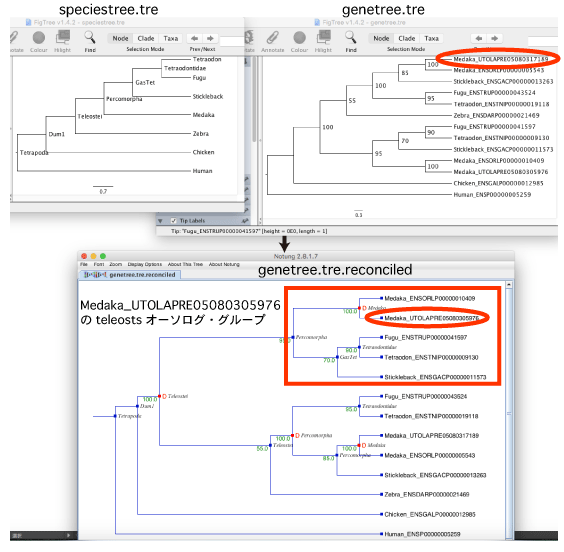
この例題は,以下のような場合を想定しています:Medaka_UTOLAPRE05080305976 をクエリとして,teleosts のオーソログを探す.
Blast hit した配列を用いて genetree.tre を推定し,Notung によって speciestree.tre と比較して Node に名前をつけます.得られた genetree.tre.reconciled をクスクリプトによって処理し,以下の条件を満たすクレードを探します.
・Medaka_UTOLAPRE05080305976 が含まれる.
・Teleostei か Percomorpha という名前でもっとも leaf が少ない.
・種分化による分岐.
([&&NHX:S=Percomorpha:Nset=<Medaka@GasTet>:H=N:D=N:B=95.0] とラベルされている)
(2016 年 11 月).
|
| Rearrange Mode (Command Line) |
|
得られた gene tree を species tree と照らし合わせて gene tree で BS 値などの支持率(Edge Weight Threshold と呼ばれています) が弱い分岐を rearrange し,その後 reconciliation を行って遺伝子重複と欠失が生じた分岐を推定します.--threthreshold オプションでブートストラップ値などの支持率の低い分岐を補正し,遺伝子重複と欠失というイベントができるだけ少なくなるような rearrangement が行われます.こちらにどのような解析が行われるか図で表されています.
Rearrange Mode は,species tree が完全二分岐である必要があります.多分岐が入っていると動きません.一方で,私はやったことがありませんが,gene tree は多分岐が入っていても解析できるようです.
command line option の例題を作りました.ブートストラップ値が 70% よりも低い分岐が補正される設定です.Reconciliation の例題と同じ 3 種類の gene tree を使っているので,結果を両者で比較することで実際にノード補正が行われているか確認することができます.
java -jar Notung-2.6.jar -s Speciestree.tre -g GRIA1.tre --rearrange --threshold 70 --info --usegenedir --speciestag prefix --log --savepng --maxtrees 5 --annotationfile 020_AnnoColor2 --homologtabletabs
-s species.tre
種の系統樹です.
-g genetree1_3R.tre
遺伝子の系統樹です.
--rearrange
threshold の値に従って,系統樹を再編します.
--threshold 70
ここでは BS 値が70% 以下の分岐を解消して再編します.ここで注意したいのが,「--threshold 70%」と書くと,得られた BS 値の最大値を 100% として,70% にあたる BS 値 (絶対値として,最大 BS 値 50 であれば, 35) 以下の枝を解消する点です (作者にメールで指摘されて,気がつきました).詳しくはマニュアル p80 「Chapter 12. Command Line Options and Batch Processing」 をご覧下さい.
--info
Duplication と loss の情報が .info ファイルに保存されます.
--usegenedir
アウトプットの tree をインファイルの tree と同じ場所に保存します.
--speciestag prefix
Species name の付け方 (e.g., Chicken_ENSGALP00000012985) を決めておきます.
--log
ntglog に解析のログ (遺伝子重複に関するパラメーターなど) が保存されます.
--savepng
PNG image として,解析の結果えられた tree を保存します.
--maxtrees
解析結果に複数の tree が出た場合に出力される最大の tree 数.
sample_rearrangeBS70.tar.gz
*sh 000_command によっても解析が始まります.
Rooting を固定
Root が 2 分岐の場合でも,アウトグループの位置が Rearrangement によって変更されてしまう場合があります.現在 rearrangement で外群を固定するオプションを付けようとしているようですが,難しいそうです (2011 年 5 月).そこで R などを使って,ingroup の根幹に BS 値 100% をつけて外群を固定して rearrangement を行います (Notung の作者に聞きました).こちらの「内群根幹の分岐に BS 100% を付ける」をご覧下さい.
|
|
Rearrange をたくさんの gene tree について一気に行うスクリプトです.
AutoNotingRear_fol.tar.gz
|
|
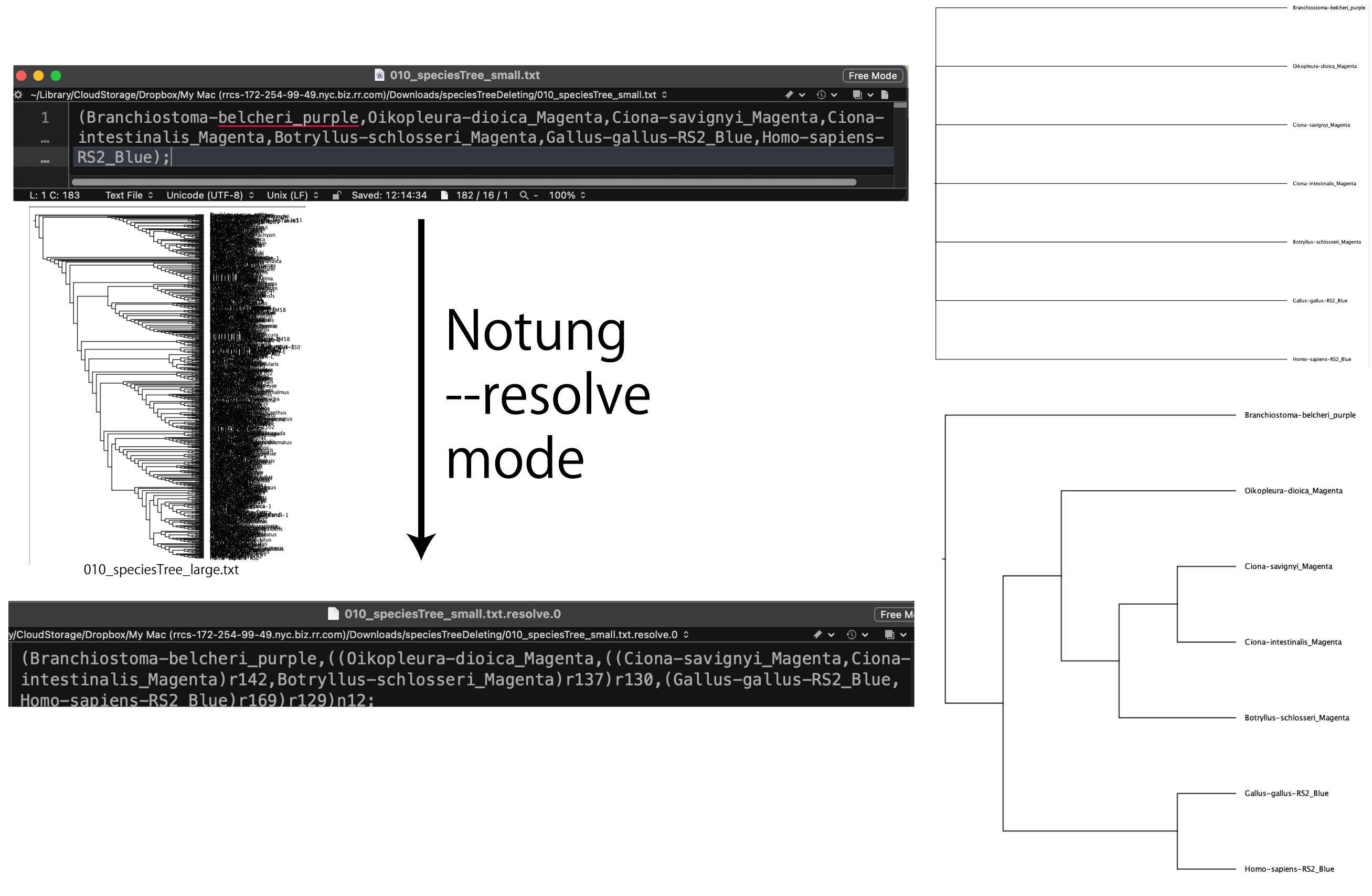
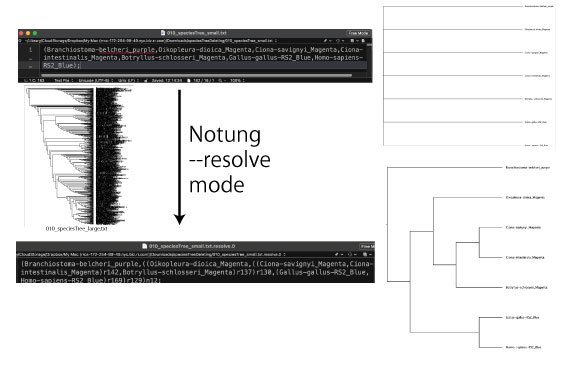 |
ポリトミーを 2 分岐にします。ここでは巨大な種系統樹から、ポリトミーで描いた種の種系統樹を抽出しています。
speciesTreeDeleting.zip (2024 年 6 月)
|
|
| NewFiles/CAMK2G_H.02a.jpg |
daughterNodeIdentifyFromNHX_PERL.tar.gz (2017 年 12 月)
daughterNodeIdentifyFromNHX_PYTHON.tar.gz (2018 年 8 月)
|
$ perl daughterNodeIdentifyFromNHX.pl
orthoNode: Euteleostomi, ENSP00000410298
6 spp.
n20[&&NHX:S=Euteleostomi:D=N:B=98.0]
DaughterNode1:
3 spp.
n14[&&NHX:S=Clupeocephala:D=N:B=100.0]
DaughterNode2:
3 spp.
n19[&&NHX:S=Tetrapoda:D=N:B=98.0]
ParentNode:
14 spp.
r65[&&NHX:S=Euteleostomi:D=Y]
|
|
| Rivera, A. S., M. S. Pankey, et al. (2010). "Gene duplication and the origins of morphological complexity in pancrustacean eyes, a genomic approach." Bmc Evolutionary Biology 10. PDF. |
|
|