|
|
| 2025 年 9 月 10 日 改訂 |
R は統計解析を行うプログラムです.無料で配布されています.一度スクリプトを書いてしまえば,条件を変えた解析を容易に行うことができます.
系統解析には Ape というパッケージが有名です.系統樹探索や年代推定など核となる解析よりも,系統樹をグラフ化したり,系統樹上で枝長を計算するなど,簡単だが手作業では難しい操作に優れています.
|
| インストール |
Mac
こちらをご覧下さい.The Comprehensive R Archive Network から最新版をダウンロードして,.dmg ファイルをダブルクリックするだけす.
アプリケーションフォルダーに「R」と「R64」という二つのアプリケーションができます.これは使っている PC のプロセッサが i386 か x86_64 によるようです.Mac であればターミナルに,
uname -p
と入力すれば,プロセッサの情報が得られます.
注意:
・バージョンが古いために関数が正常に動かないことがよくあります.そのような場合は,最新のバージョンをインストールすると問題が解決されることがよくあります.
・MacOS Big Sur で R や Rscript が Terminal から起動できないです。こちらを参考に解決しました (2021 年 4 月)。
Linux
wget https://cran.r-project.org/src/base/R-3/R-3.6.3.tar.gz
tar xzvf R-3.6.3.tar.gz
cd R-3.6.3
sudo ./configure --with-readline=no --with-x=no
sudo make
sudo make install
こちらを参照しました (2022 年ごろ)。
--with-readline=no --with-x=noは無理やりインストールなので、あまりお勧めされていないようです (link) (2024 年 7 月)。
デフォルトでは「/usr/local」にインストールされますが、ルート権限がない場合は --prefix でインストール先を変更するそうです (生物統計学) (2024 年 10 月)。
参考になるサイト
生物統計学 R インストール
遺伝研:R の使い方
Posit Community
make の途中で undefined reference to `libiconv' というエラーメッセージが出た時の対応。以下で make が進みました。
sudo apt update
sudo apt install gdebi-core
|
| アンインストール |
Complete remove and reinstall R, including all packages
$ sudo apt-get remove r-base-core
$ sudo apt-get remove r-base
$ sudo apt-get autoremove
これに加えて、以下でパッケージがインストールされているか調べることができます。これらのフォルダーを削除するようです。
$ R -e '.libPaths()'
(2024 年 7 月)
遺伝研:R の使い方
「アンインストールの方法」に、make uninstall が書かれています (2024 年 10 月)。
|
| 作業ディレクトリの設定とプログラムの実行 |
作業ディレクトリの設定
> setwd("/Users/Jun/R_fol")
作業ディレクトリの確認
> getwd()
プログラムの実行 (コマンドラインで行う場合)
> source('plot.R')
システムコマンド
> system("open .")
|
| Mac user 必見 - R 環境専用エディタ |
Mac の場合,R に入っているエディタを使うとワンタッチで動作確認ができます.
|
|
|
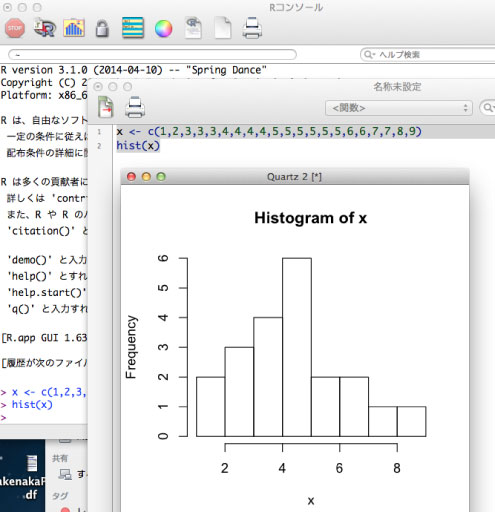
| 以下のスクリプトを Editor に貼付けて全部選択し,command+return を押すとヒストグラムが作成されます. |
x <- c(1,2,3,3,3,4,4,4,4,5,5,5,5,5,5,6,6,7,7,8,9)
hist(x)
|
|
|
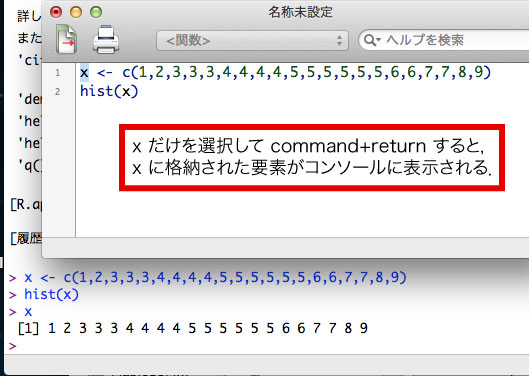
スクリプトの一部だけ動作確認することも可能です.
以上,詳しくはこちらをご覧下さい. |
 |
|
コンソールクリア
option + command + L
|
| R ヘルプ |
R ヘルプには有効なドキュメントと例題が入っています.
例えば,上記 ape を読み込んだ後で,
?plot.phylo
あるいは
help(ape)
とすることで,必要なドキュメントを見ることができます.
ape のバージョンは、
[viento vpsuser:cgi-bin]$ R
R version 3.6.0 (2019-04-26) -- "Planting of a Tree"
Copyright (C) 2019 The R Foundation for Statistical Computing
Platform: x86_64-redhat-linux-gnu (64-bit)
....
'demo()' と入力すればデモをみることができます。
'help()' とすればオンラインヘルプが出ます。
'help.start()' で HTML ブラウザによるヘルプがみられます。
'q()' と入力すれば R を終了します。
> library("ape")
> packageVersion("ape")
[1] ‘5.6.2’
>
で調べることができます。
|
| ape パッケージのインストール |
ape は分子系統解析のパッケージです.通常は,R のコンソールに以下を打ち込めばインストールされます.
install.packages("ape")
詳しくはこちらを参照してください.
R の最新版に上記の手順で ape がインストールできないことがありました.そこで root で以下の手順に従いソースコードからインストールしました.
1. こちらから ape_3.3.tar.gz をダウンロード.
wget http://cran.r-project.org/src/contrib/ape_3.3.tar.gz
2. コンパイル
R CMD INSTALL ape_3.3.tar.gz
こちらの Linux の箇所を参照しました (2015 年 6 月).
ミラーサイトの変更
CentOS7 で ape がインストールできない場合がありました。まず、サーバーで、sudo を使って R を立ち上げました:
sudo /usr/local/bin/R
その後、ape のインストールを試しました:
install.packages("ape")
すると、以下のエラーメッセージが R から出ました。
URL 'https://cran.ism.ac.jp/src/contrib/PACKAGES' を開けません
ミラーサイトを変更したら無事インストールできました。インストールの途中でミラーサイトを JAPAN にすると、統計数理研究所が選ばれます。この時は、 統数研のサーバーに何らかの問題があったようです。
R に以下を入力して、再度、install.packages("ape") を行います。
options(repos="https://ftp.yz.yamagata-u.ac.jp/pub/cran/")
こちらを参照しました (2021 年 3 月)。
ライブラリを指定してパッケージをインストール
web tool などの場合、Rscript などから R を操作するので、ユーザーが apache になります。この場合、パッケージのインストールする場合、場所を指定する必要があります。そうしないと、terminal からの操作では ape が使えますが、web tool (apache) からの操作では ape が使えないことがあります。 apache が R を立ち上げるので、該当するライブラリに ape が入っている必要があるのです。
まず、sudoコマンドを用いて、terminal から R を立ち上げます。
sudo R
その後、R のコンソールから以下の操作を行います。
library()
以下のような表示が出ます。ライブラリ ‘/usr/lib64/R/library’ に ape が入っていませんね。
パッケージ (ライブラリ ‘/home/jinoue/R/x86_64-redhat-linux-gnu-library/3.6’ 中):
ape Analyses of Phylogenetics and Evolution
Rcpp Seamless R and C++ Integration
パッケージ (ライブラリ ‘/usr/lib64/R/library’ 中):
base The R Base Package
boot Bootstrap Functions (Originally by Angelo Canty
for S)
....
以下のコマンドによって、ライブラリ ‘/usr/lib64/R/library’ に ape をインストールします。
install.packages("ape",lib="/usr/lib64/R/library")
インストールできたか確認してみます。
library()
パッケージ (ライブラリ ‘/usr/lib64/R/library’ 中):
ape Analyses of Phylogenetics and Evolution
base The R Base Package
boot Bootstrap Functions (Originally by Angelo Canty
for S)
....
こちらを参照しました。ありがとうございました (2020 年 5 月)。
|
| 基本的なテクニック |
R コンソールをクリアにする
option + command + l
ターミナルの「コマンド+K」にあたります.
スクリプトを途中で止める
q()
です.perl の「exit;」 に相当します.
作業ディレクトリの内容を見る
dir()
作業ディレクトリのアドレスを表示する
getwd()
作業ディレクトリを変更する
setwd('/Users/JunInoue/Downloads/histR')
ファイルを読み込んでデータフレームを作成する.
read.table('xxx.txt', header = T, sep = '\t') とすると,データが欠けている場合 (例: エクセルで空欄) は NA と認識.詳しくは竹中先生のページ「補足: 結束値などの扱い」を参照してください.
read.table()
read.table でデータを df と入力して読み込んだ後に,因子になってしまった変数を文字列に変換する.こちらを参照しました.
df$ID<- as.character(df$ID)
データの変数名をリストアップする
names()
要素の種類をリストアップする
unique()
データ内にある変数の種類を確認する
str()
変数の確認
ls() または objects()
解析結果を .dat ファイルとして保存する
save(aValues, bValues, nValues, file="tancha.dat")
.dat ファイルを読み込む
load("tancha.dat")
グラフをPDF ファイルとして保存する
dev.copy2pdf(out.type="pdf",file="tancha.pdf")
データ内にある変数の種類を確認するパターンマッチ grep regexpr.こちらを参考にしました.ここでは files ベクトル内部で ".gb"で終わる要素を抽出しています.
grep("\\.gb$", files)
日本語の文字化け
plot した図で文字化けする場合は,こちらを参考に処置してください.
|
| 2 つのデータフレームの列を比較して統合する |
| mergeDF.tar.gz (2013 年 5 月) |
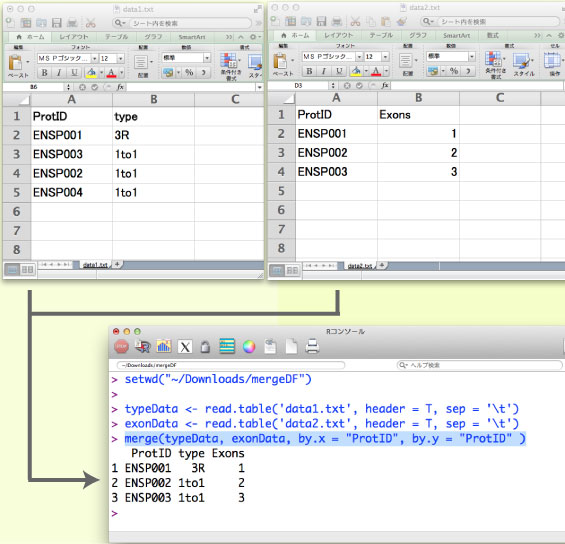
|
| グラフ作成例 |
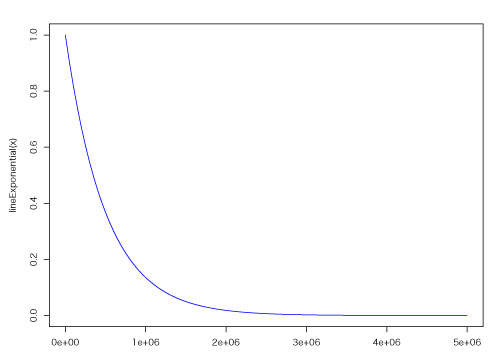
対数減数曲線
例題として,WGD 由来重複遺伝子の対数減数モデルの曲線を描きます.
Force et al. (1999) は Nei and Roychoudhury (1973) が使っている対数曲線の式に基づいて,重複遺伝子の機能が失われる確率を「1-e^(-2ut)」と提唱しています.t は世代時間,u は null mutation rate です.t を時間と考えると,ここで用いられた e^(-2ut) は重複遺伝子の欠失率をモデル化した式と見なすことができます.この式,
y= e^(-2ut)
を R を使ってグラフ化してみましょう.突然変異率 u は 1E-6 とします.
1E-6
は指数標記で 1*10^(-6) と同じです.
## グラフをプロットします.xlim = c(0, 5e+6) は表示される x 軸の範囲です.
lineExponential = function(x) exp(-2*(1E-6)*x)
plot (lineExponential, xlim = c(0, 5E+6), 0,col = "blue")
## WGD が生じた時点 x=0 の残存率は 1 となっていることを確認します.
x=0
exp(-2*(1E-6)*x)
## 100 万年後に残っている割合を見てみましょう.0.135... になっています.
x=1E+6
exp(-2*1E-6*x)
|
|
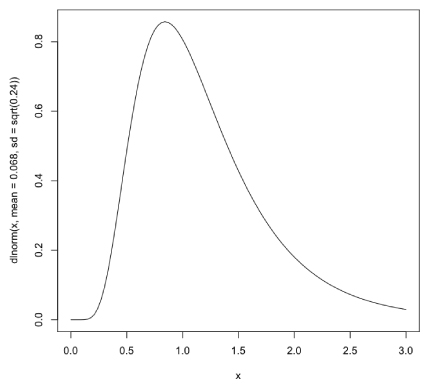
対数正規分布 log-normal distribution
x <- seq(0, 3, 0.05)
plot(x,dlnorm(x, mean=0.068, sd=sqrt(0.24)), type="n")
curve(dlnorm(x, mean=0.068, sd=sqrt(0.24)), type="l",add=T)
以下のようなカーブが描けるはずです.
こちらを参考にしました.Log-noarmal distribution についてはこちらを参考にしてください.
|
| 簡単な散布図の作成 |
こちらを参考にして,5 分で例題からプロットができるようになりました.
もっとも簡単な操作を以下に書きます.
1.
作業ディレクトリの設定 (Mac の場合)
メニューバーから,
R > 環境設定 > 起動
を選び,初期作業ディレクトリを設定します.常に適用をチェックすると,選んだディレクトリが毎回作業ディレクトリとして選ばれます.
2.
オブジェクトの作成
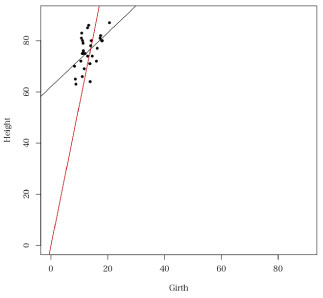
testtrees <- read.table("trees.txt", header = TRUE)
3.
プロットの作成
plot(Height ~ Girth, data = testtrees)
メモリの範囲を調節したり,プロットを変更することもできます.
plot(Height ~ Girth, data = testtrees,xlim = c(0,90),ylim = c(0,90),pch = 20)
3.
回帰分析
result2 は原点を通る直線回帰です.こちらを参考にしました.
result <- lm(Height ~ Girth,data=trees)
result2 <- lm(Height ~ Girth-1,data=trees)
推定回帰直線を描きます.
abline(result)
abline(result2)
分析結果の要約を出します
summary(result)
以下です.1.0544 が傾きです
Call:
lm(formula = Height ~ Girth, data = trees)
Residuals:
Min 1Q Median 3Q Max
-12.5816 -2.7686 0.3163 2.4728 9.9456
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) 62.0313 4.3833 14.152 1.49e-14 ***
Girth 1.0544 0.3222 3.272 0.00276 **
---
Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1
Residual standard error: 5.538 on 29 degrees of freedom
Multiple R-squared: 0.2697, Adjusted R-squared: 0.2445
F-statistic: 10.71 on 1 and 29 DF, p-value: 0.002758
4. 相関係数
相関係数 R を出します.
cor.test(testtrees$Height,testtrees$Girth)
出力です.
Pearson's product-moment correlation
data: testtrees$Height and testtrees$Girth
t = 3.2722, df = 29, p-value = 0.002758
alternative hypothesis: true correlation is not equal to 0
95 percent confidence interval:
0.2021327 0.7378538
sample estimates:
cor
0.5192801
testtrees$Height と testtrees$Girth の相関係数は 0.5192801 です.
散布図と相関係数を得る
こちらを参考にして,散布図の作成をターミナルから行うようにしました.rscript というプログラムを使います.rscript については こちらを参照してください.
実は rscript だけでなく,Rscript というおのもあって,これは Mac でも Linux でも使えます.Linux では rscript はインストールされていませんでした.「?Rscript」で解説を読んでください.
rsScatterTime_fol.tar.gz を解凍して,ターミナルで作成されたフォルダに入ってください.
rscript scatterTime.R PosMean.txt
とタイプすると,散布図と相関係数が書かれた PDF ファイルが得られます.以下がスクリプトです.
#### rscript scatterTime.R PosMean.txt
### ファイル名 (PosMean.txt) をデータフレームに入れて,ファイルを読み込む
args <- commandArgs() # パラメータのベクトル
data.file <- args[6] # 6番めの要素が最初のパラメータ
d <- read.table(data.file, header = T)
d.act <- d[42 <= d$node & d$node <= 50,]
d.sar <- d[51 <= d$node,]
d.oth <- d[d$node <= 41,]
### スクリーンアウト
cat("inputFile: ")
cat(data.file)
cat("\n")
##### Main program ####
### デバイスの用意
## アウトファイル・データフレームの作成
image.file <- paste(data.file, ".pdf", sep = "")
pdf(image.file)
### プロット
# 枠の作成
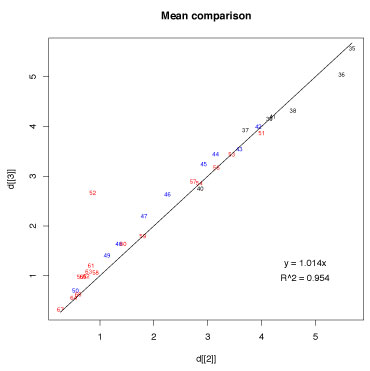
plot(d[[2]], d[[3]], type="n", main = "Mean comparison", xlim = range(d[2]), ylim = range(d[3]))
## node number をプロットする場合
text(d.act[[2]], d.act[[3]], label=d.act[[1]], cex = 0.7, col='blue')
text(d.sar[[2]], d.sar[[3]], label=d.sar[[1]], cex = 0.7, col='red')
text(d.oth[[2]], d.oth[[3]], label=d.oth[[1]], cex = 0.7, col='black')
## 点でプロットする場合
#par(new = T)
#plot(d.act[[2]], d.act[[3]], pch = 20, xlab = '', ylab = '', xlim = range(d[2]), ylim = range(d[3]), col='blue')
#par(new = T)
#plot(d.sar[[2]], d.sar[[3]], pch = 20, xlab = '', ylab = '', xlim = range(d[2]), ylim = range(d[3]), col='red')
#par(new = T)
#plot(d.oth[[2]], d.oth[[3]], pch = 20, xlab = '', ylab = '', xlim = range(d[2]), ylim = range(d[3]), col='black')
### 原点を通る回帰直線の傾きを求めて,図に回帰曲線を引く
lf <- glm(d[[3]] ~ d[[2]] - 1) # モデルのあてはめ結果を lf にしまう.
cf <- lf$coefficients
curve(x + 0,to = max(d[[2]]), add= T)
eq <- sprintf('y = %.3fx',cf[1])
### 重相関係数を求める
c.result <- cor.test(d[[2]], d[[3]])
r <- c.result$estimate
r2 <- r^2
Rsq <- sprintf("R^2 = %.3f", r2)
## 回帰直線の式を書き込む
# 現在の座標系を取得
par('usr') -> usr
# x軸の,左から 80% のところ.
x <- (usr[1] + usr[2]) * 0.8 + usr[1]
# y軸の,下から 20% のところ.
y <- (usr[4] - usr[3]) * 0.2 + usr[3]
text(x, y, eq)
# 重相関係数を書き込む
y <- (usr[4] - usr[3]) * 0.15 + usr[3] # y軸の,下から 15% のところ.
text(x, y, Rsq)
## デバイスを閉じる
dev.off()
|
| 二項分布の P 値 |
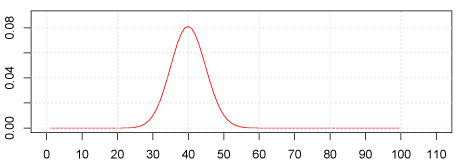
確率変数 X が 二項分布 B(100, 0.4) に従うとき,X が 50 以上となる確率,P 値,を求めます.
参考にしたページ
http://cse.naro.affrc.go.jp/takezawa/r-tips/r/60.html
https://oku.edu.mie-u.ac.jp/~okumura/stat/tests_and_CI.html
|
##
## B(100, 0.4) の二項分布で,50 の P 値を求める
obs <- 50
total <- 100
rate <- 0.4
### 手計算
ans <- 0
ansD <- NULL
for (i in obs:total) {
## choose は二項係数を計算する
ansTMP <- choose(total, i)* rate^i * (1-rate)^(total-i)
# ansTMP <- ifelse(is.nan(ansTMP), 0, ansTMP)
ans <- ans + ansTMP
ansD <- c(ansD, ansTMP)
}
ans
### pbinom () 関数で計算.lower.tail=FALSE で右側の推定.詳しくは ?rbinom
pbinom(obs-1, total, rate, lower.tail=FALSE)
###### PDF drawing
### 1〜total
par(ann=F)
plot(1:total, dbinom(1: total, total, p=rate), type="l",
xaxp = c(0, 110, 11), xlim=c(0, 110), ylim=c(0, 0.09), col=2)
grid()
### obs-10〜
#par(ann=F)
#plot(obs:total, dbinom(obs: total, total, p=rate), type="l",
# xaxp = c(obs-10, 100, 6), xlim=c(obs-10, 100), ylim=c(0, 0.09), col=2)
#grid() |
|
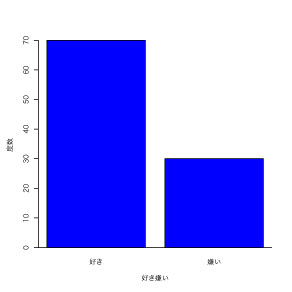
| 棒グラフの作成 |
こちらをご覧下さい.
freq <- c(70,30)
names(freq) <- c("好き","嫌い")
barplot(freq, col="blue", xlab="好き嫌い", ylab="度数")
abline(h=0)
をテキストファイルとして保存します.メニューバーのファイル/ソースを読み込むを選んで,上記のテキストファイルを読み込みます. |
 |
|
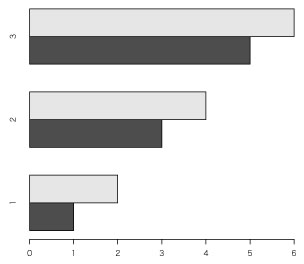
a <- c(1, 3, 5)
b <- c(2, 4, 6)
#x <- matrix(c(1, 2, 3, 4, 5, 6), 2, 3)
x <- rbind(a, b)
x
barplot(x, horiz=T, beside=T, names=1:3) # Horizontal
#barplot(x, horiz=T, names=1:3) # Horizontal bar
|
|
| ヒストグラムの作成 |
例題 1
ヒストグラムを作成します.?hist と入力して,最後の例題を見てみましょう.R にはすでに例題が入っていて,
hist(islands)
とすることで,ヒストグラムを作成してくれます.例えば,x というベクトルを作成して,
x <- c(1,1,2,3,4,4,5)
hist(x)
としても,ヒストグラムが作成されます.こちらに詳しい説明があります.
最頻値
hist 関数を使いますが,plot させません.
xHist <- hist(x, plot=FALSE)
which(xHist$counts==max(xHist$counts))
こちらを参照しました (2013 年 4 月).
|
|
例題 2
.histR.tar.gz
ダウンロードして得られた histSimple.R を R アイコンに ドラッグ&ドロップして R のエディターで開きます.コマンド+ A で全部を選択した後,コマンド+リターンで R を走らせます. |
 |
|
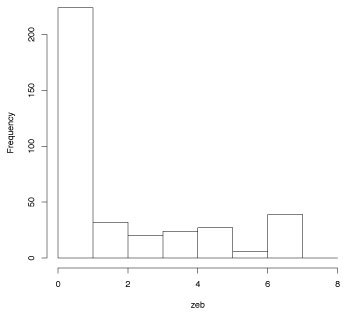
例題 3
1000a.dat.tar.gz
.dat として保存されたデータを読み込んでヒストグラムを書きます (2015 年 6 月).
> cc <- load("~/.../1000a.dat")
> str(cc)
chr [1:4] "aValues" "bValues" "dValues" "nValues"
> aValues
[1] 0.0025884912 0.0023656549 ....
[1000] 0.0026137924
> median(log(aValues))
[1] -5.964303
> hist(log(aValues),breaks=50)
|
 |
複数のヒストグラムを一枚の図にまとめる
histMultipleExons.tar.gz
図の並べ方に関しては,こちらを参照しました (2013 年 4 月).
|
 |
連続変数からサンプリングした値を階級ごとに集計 |
>Z <- stats::rnorm(10000)
>table(cut(Z, breaks = -6:6))
>as.integer(table(cut(Z, breaks = -6:6)))
こちらを参照しています.意外とこのテクニックは見つかりません.サイトがなくなったときのために,コマンドだけ移させていただいています.
|
| Lattice を使ってヒストグラムを描く |
Lattice (ラティス) パッケージを使うと,複数のグラフを一枚の図として並べるなど,R を使ったグラフィックスがさらに便利になります.
install.packages("lattice")
によって lattice パッケージをインストールしてください.その後,ダウンロードして得られた histLattice.R を走らせると,以下のような図が得られるはずです.
histLatticeR.tar.gz
|
|
d <- read.table(file="infile.txt",header = TRUE)
### Retrieve the recoreds with distance for less than 2
d_less2 <- d[d$Dist < 2,]
################## Make histograms
pdf("histLattice.pdf")
library(lattice)
histogram( ~ Dist | Class, data = d_less2,
layout = c(1,3),nint = 30, xlab = "Distance", strip = FALSE,
strip.left = TRUE, ylab="Frequencies")
dev.off()
################## Calcurate the averages of distances
## Check the variables
names(d_less2)
unique(d_less2$Class)
## Retrieve the lines of each class
typeR <- d_less2[d_less2$Class == "typeR",]
typeM <- d_less2[d_less2$Class == "typeM",]
typeO <- d_less2[d_less2$Class == "typeO",]
## Calculate the average of each class
mean(typeR$Dist)
mean(typeM$Dist)
mean(typeO$Dist) |
|
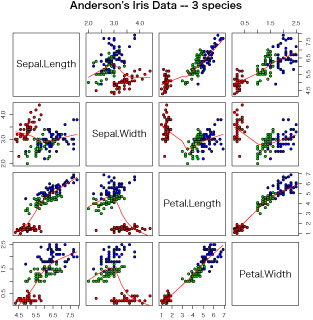
| 対散布図 |
いくつか存在する変数の相互関係を考察するには,総当たりで散布図を表示する対散布図が便利です.以下の行をコピーして R のコンソールに貼付けると,下に示した図が得られます.
pairs (iris[1:4], main = "Anderson's Iris Data -- 3 species", pch = 21, bg = c("red", "green3", "blue")[unclass(iris$Species)], panel=panel.smooth)
iris[1:4]
iris というデータはすでにデータフレームとして R に組み込まれています.
bg = c("red", "green3", "blue")[unclass(iris$Species)],
種ごとに色分けをしています.Species というコラムには 3 種があります.「iris」 と打ち込んで,データフレームを確認してください.
panel=panel.smooth
散布図の点の傾向を示す曲線が描かれます.
詳しくは,コンソールに「?pairs」と打ち込むと得られる説明を読んでください.
|
|
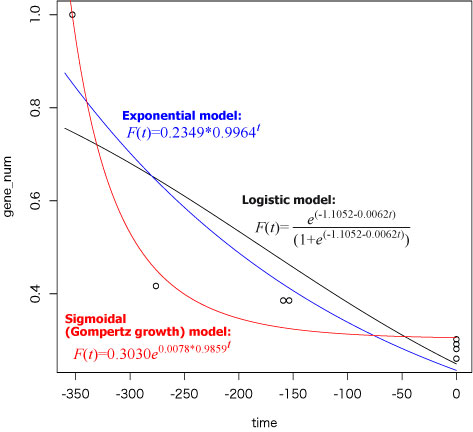
| 非線形回帰分析 A nonlinear regression analysis |
nls, glm 関数
ある分布のプロットを,ロジスティック関数 (logistic model) ,指数関数 (exponential model),左右非対称の S 字曲線の関数 (sigmoidal model) へ回帰します.長いのでこちらのページに移動しました [2013 年 1 月].
|
|
nlmrt パッケージ
nlmrt 関数は非線形最少二乗問題に信頼性の高い結果を提供することを目的としたパッケージです.非線形回帰分析を行うには nlxb という関数を用います.非線形回帰分析によく用いられている nls 関数は,残差 (residual) が小さいか,あるいはない場合には解が得られません.さらに,解析の際に与える starting parameters が実際の解と大きく異なるために,解が得られないことがよくあります.nlmrt はこれらの問題を解決するよう作られているようです.
ただし,nls と違って nlxb で得られる解 (リスト ?) は関数 AIC( ) には対応していません.AIC( ) の解を得る場合は,nlxb で得られたパラエータを用いて一度 nls で計算して,得られた解で AIC を算出する必要があります.
詳しくは以下を参照してください.
Package 'nlmrt'
nlmrt-vignette |
| 例題です.nlmrtTEST.R.tar.gz |
library(nlmrt)
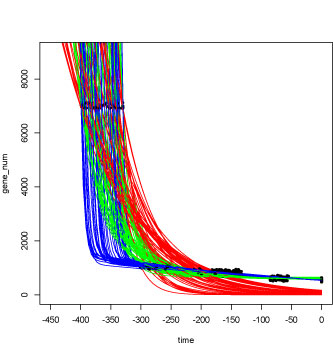
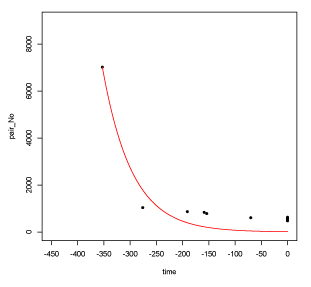
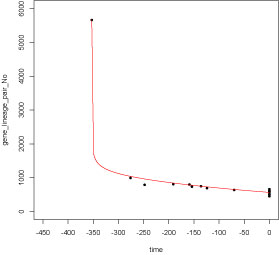
time = c(-353.0, -276, -191, -159, -154, -70, 0, 0, 0, 0, 0, 0)
gene_num = c( 7019, 1040, 868, 839, 786, 610, 630, 505, 563, 577, 476, 551)
threeRxTime <- time[[1]]
plot(time, gene_num, xaxp = c(-450, 0, 9),
xlim=c(-450,0), pch=20, ylim=c(0,9000))
par(new=T)
##################################################################
# Double exponential:
#F(t)=a *e^(-b *(t+353))+c *e^(-d*(t+353))
#F(t)=5739*e^(-0.448*(t+353))+1280*e^(-0.00237*(t+353))
#
<- Rough estimation by manual
dat <- data.frame(n=gene_num, t=time)
doubleExponential <- nlxb( n~a*exp(-b*(t-threeRxTime))+
c*exp(-d*(t-threeRxTime))+e,
data=dat, start=c(a=6000,b=0.01,c=1000,d=0.002,e=100), lower=0,
upper=c(a=10000,b=0.1,c=10000,d=0.1,e=10000))
va <- doubleExponential$coefficients[["a"]]
vb <- doubleExponential$coefficients[["b"]]
vc <- doubleExponential$coefficients[["c"]]
vd <- doubleExponential$coefficients[["d"]]
ve <- doubleExponential$coefficients[["e"]]
linearDoubleExp <- function(x) va*exp(-vb*(x-time[[1]]))
+ vc*exp(-vd*(x-time[[1]]))+ve
plot (linearDoubleExp, -450, 0, add=TRUE, col ="blue")
doubleExponentialNls <- nls(gene_num ~ va*exp(-vb*(time-time[[1]]))
+ vc*exp(-d*(time-time[[1]]))+ve, start=list(d=0.001))
AIC(doubleExponentialNls)
|
|
|
|
参考文献
回帰分析入門
とてもわかりやすいです [2013 年 12 月].
最適化問題
optim()/optimize() 関数について [2014 年 10 月].
|
関数の値を最小にするパラメータの推定 |
optim を使うことによって,ある関数の値を最小にするパラメータを選べます.以下では,最小二乗法で用いる残差の平方和を最小にしています.
まずは,こちらのページを見た方が良いです.
続いて発展として,関数が複雑に連携した場合を想定して,パラメータを関数から関数へと渡して,関数を最小にするパラメータを探すプログラムを作成しました.control をうまく設定すると,より精度が増すそうです.leastSquares_multiFunc.R.tar.gz
参考にしたサイト:
R の関数 optim の使い方
関数の最大・最小化
最小二乗法 (ウィキペディア)
Least squares (Wiki)
|
| ターミナルから R を動かす 1 |
バッチモードです.ターミナルから R を動かすと,perl などと組み合わせて使うことができます.以下をダウンロードして得られたフォルダに入って,
R --vanilla --quiet --args in.tre < script.R > log
と入力してください.Tree が書かれた 2 つのファイル (in.tre.png, in.tre.nwk) が得られるはずです.script.R がバッチファイルです.log はスクリーンアウトです.
batchR.tar.gz
こちらを参照しました.
|
# Load package ape.
library(ape)
# Read files
args <- commandArgs()
treeFile <- args[5] # in.tre
# Read tree
tr <- read.tree(treeFile)
# Open the device
png.file <- paste(treeFile, ".png", sep = "")
# Plot tree
png(png.file)
plot.phylo(tr)
# Save the tree as the newick format
nwk.file <- paste(treeFile, ".nwk", sep = "")
write.tree(tr, file=nwk.file)
dev.off()
|
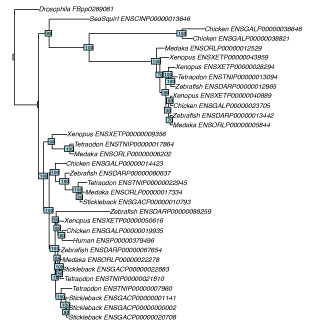
| ターミナルから R を動かす 2: rscript |
rscript というプログラムを使って,R をターミナルから動かすこともできます.rscript についてはこちらを参照してください.ここでは rscript を使って newick tree を PDF ファイルに保存します.
newickTreePlot_fol.tar.gz をダウンロード・解凍し、これに terminal で入ってください。そして,
rscript plotNewick.R MyTree.tre
と入力してください.この場合,R が使っている PC にインストールされている必要があります.解析がうまく動けば,Rplots.pdf というファイルに以下の tree が保存されます.R と系統樹, 1, 2,を参考に作成しました.
注意: スクリプトを他の PC で試すと,以下のようなエラーメッセージが出て,rscript が動かないことがよくあります.その場合は,まず rscript を入れ替えてみましょう. rscript と R のバージョンがあわないと動かないです.
[inouejun:newickTreePlot_fol]$ rscript plotNewick.R MyTree.tre
Rscript execution error: No such file or directory
[2018 年 2 月].
|
 |
# ape を読み込む
library(ape)
# newick format のファイルを読み込む
#MyTree <- read.tree("MyTree.tre")
# ファイル名 (PosMean.txt) をデータフレームに入れて,ファイルを読み込む
args <- commandArgs() # パラメータのベクトル
MyTree <- args[6] # 6番めの要素が最初のパラメータ.
MyTree <- read.tree(MyTree)
### 枝長がマイナスになる場合は、0 に置き換える。
MyTree$edge.length[MyTree$edge.length<0]<-0
# 系統樹を書いたときに,上にルートが来るようにする
MyTree <- ladderize(MyTree)
# Tree をプロットする.
plot.phylo(MyTree,no.margin = TRUE, font = 3,cex = 0.8)
# BS 値をプロットする.
rs1 <- round(runif(13, 60, 100), 0)
nodelabels(MyTree$node.label,cex=0.55)
#dev.off()
|
| 外れ値の検出-1 |
スミルノフ・グラブス検定 (Smirnov-Grubbs test) によって外れ値を検出するスクリプトを書きました.こちらのサイトを参考に作成しました [2011 年 11 月].
sg.R.tar.gz
|

|
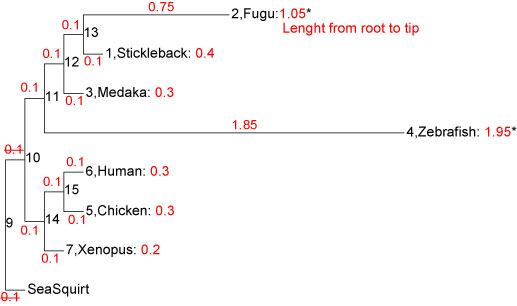
| 外れ値の検出-2: long branch |
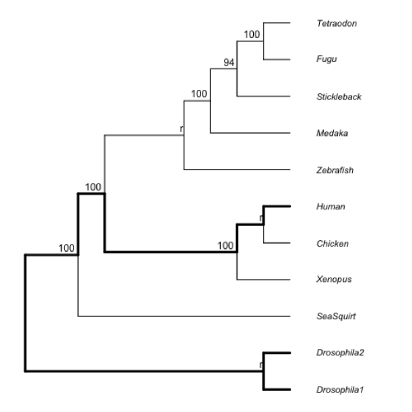
スミルノフ・グラブス検定を用いて,root to tip length (ここでは Node10 から末端まで) が平均値から極端に異なる枝 (以下の図で * ) を検出するプログラムです.外群 (SeqSquirt) は検定から外されます.もちろん可能であれば相対速度検定を行うべきですが,計算が思ったほど簡単ではないので,ここではスミルノフ・グラブス検定を用いています.しかし,この検定方法が長枝の検出に向いてているかどうかは,検討が必要です.ご存知の方がいたら教えてください [2012 年 2 月].
SGforBL.tar.gz
両側検定なので、p 値を計算する際の自由度は n-2 にしているようです。こちらを参照しました [2020 年 1 月]。
|
|
|
|
## This program returns the data frame including root to tip branch lengths
library(ape)
mytree = read.tree(text = "(((((Stickleback:0.1,Fugu:0.75)68:0.1,Medaka:0.1)93:0.1,
Zebrafish:1.85)90:0.1,((Chicken:0.1,Human:0.1)92:0.1,
Xenopus:0.1)87:0.1)99:0.1,SeaSquirt:0.1)100;")
br.rootTip <- function(myTree)
{
myTree$node.label <- ((length(myTree$tip)+1):((length(myTree$tip)*2)-1))
distRtoTs <- c() # branch lengths from root to tip
names <- c() # leaf names
for(i in 1:length(myTree$tip.label)-1) # -1 deletes the outgroup
{
names <- c(names,myTree$tip.label[i])
# Calculate the branch lengths and push distances
# into the vector, distRtoTs
distRtoT <- dist.nodes(myTree)[(length(myTree$tip)+2),i]
distRtoTs <- c(distRtoTs, distRtoT)
# Change leaf names
myTree$tip.label[i] <- paste(i,',',myTree$tip.label[i],':', distRtoT, sep="")
}
# Make the data frame [leaf, dist]
Dfrm <- data.frame(NAMES = names, DIST = distRtoTs)
return(Dfrm)
}
## Calculate the branch length from root to tip
nameLengthDfrm <- br.rootTip(mytree)
nameLengthDfrm
|
|
newick tree 状で leaf 間の距離を計算するには,ape の cophenetic 関数を使います cophenetic.R.tar.gz.こちらを参照しました [2012 年 2 月].このサイトを参照して,こちらに Root to tip 距離を計算する CGI スクリプトを作ってみました.
cophenetic(geotree)["pallida", "conirostris"]
|
| ランダムな系統樹を作成する |
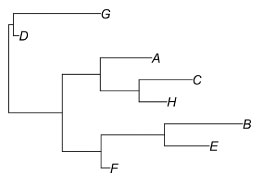 |
library(ape)
myTree1 <- read.tree(text = "(A,B,C,D,E,F,G,H);")
plot(rtree(8, rooted = TRUE, tip.label = myTree1$tip.label))
(2024 年 6 月)
|
|
## Set the working directory. Type "pwd" in terminal and get the address.
setwd("/Users/inoue_server/Desktop/blOne2one")
## Invoke the library APE
library(ape)
## Read the newick tree file
tr <- read.tree("RAxML.tre")
## Calculate the branch lengths betwee the two leaves
humZeb <- cophenetic(tr)["Human_ENSP00000301740", "Zebrafish_ENSDARP00000106523"]
humCod <- cophenetic(tr)["Human_ENSP00000301740", "Cod_ENSGMOP00000016088"]
humFly <- cophenetic(tr)["Human_ENSP00000301740", "Drosophila_FBpp0292226"]
## Just printout the calculated branch lengths
humZeb
humCod
humFly
|
|
FigTree で確認すると以下のようになります.
|
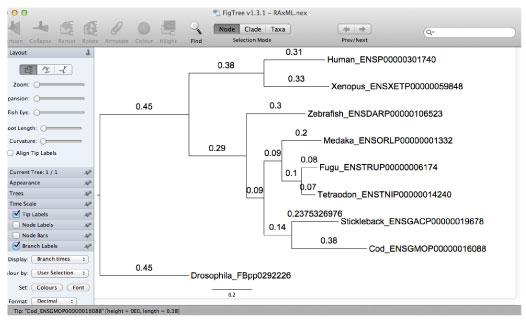 |
blOne2one.tar.gz
|
| あるノードから末端までの枝長を求める |
| nodeTipBL.tar.gz |
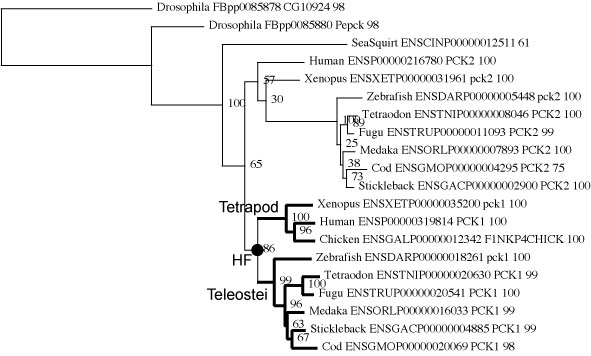 |
ダウンロード・解凍して得られた calNodeToTip.R ファイルを R で読み込み,コマンドを実行 (Mac であれば,command + リターン) してください.以下のような 3 つの値が out_nodeTipBL ファイルに追加されてゆきます.
>BLcompHF
0.1592556: HF ノードから Teleostei クレード各末端までの平均枝長 - (1)
0.1656951: HF ノードから Tetrapod クレード各末端までの平均枝長 - (2)
1.040435: (2) / (1)
|
| Package のインストール |
例えば,系統解析で有名な ape という package をインストールするには,インターネットがつながって PC から
install.packages("ape")
と入力してください.あとは画面の指示に従ってください.その後,
library(ape)
によって,ape が読み込まれて使える状態になります.library (ape) は毎回行う必要があります.プログラムであれば,毎回最初の方に書いておく必要があります.
詳しくはこちらを参照してください.
|
| ape: newick tree を描く |
ape というパッケージを使います.
install.packages("ape")
と入力して ape をインストールしてください.
drewTree_fol.tar.gz を解凍して,得られた drawTree.R と Mytree.tre を自分の作業ディレクトリーに入れてください.R のコンソールから、
setwd("~/Downloads/drawTree_fol")
と入力して、ディレクトリーに移動します。その後、
source('drawTree.R')
り入力して drawTree.R を R で読み込むと,以下のような図が得られます (MyTree.tre.pdf ファイル).

# ape を読み込む
library(ape)
# インファイル名を変数に格納する
inFile.name <- ("MyTree.tre")
# tree をインファイルから読み込む
MyTree <- read.tree(inFile.name)
# プリントアウトに必要なデバイスを用意する.
image.file <- paste(inFile.name, ".pdf", sep = "")
pdf(image.file)
# 系統樹を書いたときに,上にルートが来るようにする
MyTree <- ladderize(MyTree)
# 系統樹を書いたときに,下にルートが来るようにする
#MyTree <- ladderize(MyTree,FALSE)
# Tree をプロットする.
plot(MyTree)
# newick tree を nexus 形式で保存する.
#write.tree(MyTree, file="MyNewickTreefile.tre") # Phylip 形式.
write.nexus(MyTree, file="MyNexusTreefile.nex") # Nexus 形
# dev.off という関数でデバイスを閉じます.
dev.off()
|
| PDF: en-dash 問題 |
library(ape)
myTree1 <- read.tree(text = "(A-1,(B-1,(C-1,D-1)));")
cairo_pdf("output_cairo.pdf")
plot(myTree1)
dev.off()
pdf("output_normal.pdf")
plot(myTree1)
dev.off()
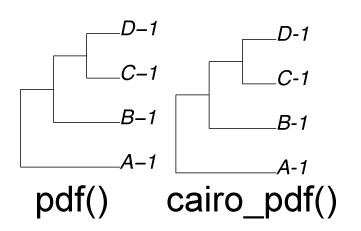
2026 年 6 月。
|
| Negative branch length を 0 にする |
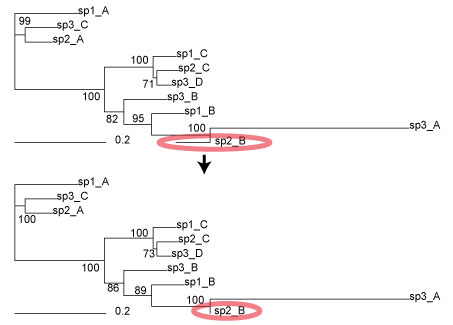 |
Gene_tree$edge.length[Gene_tree$edge.length<0]<-0
negativeBranchLengthR.tar.gz
R のスクリプトはこちらを参照しました.この操作に関しては,こちらのサイトでは,Kuhner and Felsenstein (1994) が参考文献として挙げられています.
NJ 法によって系統樹を推定すると,branch length がマイナスになることがあります.この,negative branch length を含んだ newick format を描画すると,上の tree のように,枝が反対側に出てしまいます.
Mega や TreeView X などでは,描画してみると,これら negative branch length を 0 にしているようです (参考文献は見つけられませんでした).Negative branch length 問題に関しては,Salemi et al. (2009, P302) でも言及されています.
AS さん,情報ありがとうございます (2019 年 2 月).
|
| ape: node number を確認する |
ape で系統樹を解析するとき,node に番号が振られます.以下 (nodeLabeler.R.tar.gz) のコマンドを使えば,node 番号を確認することができます.こちらを参照しました.
|
library(ape)
#inFile.name <- ("treeNwk.tre")
#myTree <- read.tree(inFile.name)
myTree <- read.tree(text = "(((((A,B),C),D),((E,F),G)),H);")
myTree$node.label <- ((length(myTree$tip)+1):((length(myTree$tip)*2)-1))
for(i in 1:length(myTree$tip)){
cat(i,myTree$tip[i],'\n')
}
plot(myTree, show.tip.label=TRUE, show.node.label=TRUE, no.margin=TRUE, )
|
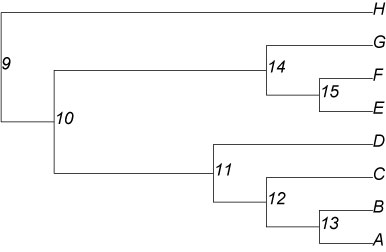
|
| ape: reroot |
rerootR_fol.tar.gz をダウンロード・解凍した後にできたファイルに入って,
source('reroot.R')
と入力してください.MyTree.trererooted.pdf というファイルにアウトファイルが保存されます.
# source('reroot.R')
# ape を読み込む
library(ape)
# インファイル名を変数に格納する
inFile.name <- ("MyTree.tre")
# アウトグループを設定する
outGroup <- "Human"
# tree をインファイルから読み込む
MyTree <- read.tree(inFile.name)
# プリントアウトに必要なデバイスを用意する.
image.file <- paste(inFile.name, "rerooted.pdf", sep = "")
pdf(image.file)
# 一つの PDF ファイルに二つの tree を並べる
layout(matrix(1:2,1,2))
# 系統樹を書いたときに,上にルートが来るようにする
# MyTree <- ladderize(MyTree,FALSE)
MyTree <- ladderize(MyTree)
## 樹長なしで Tree をプロットする.
plot(MyTree,use.edge.length = FALSE)
# BS 値をのせる
#nodelabels(t$node.label)
## 実は root では根幹 3 分岐になってしまう.
# 試しに,下にあるように is.rooted(rerooted) と打つと,
#FALSE と帰ってくる.
# rerooted <- root(MyTree,"Human")
# is.rooted(rerooted)
## そこで,r=T オプションを使うと,根幹 2 分岐になる.
# PDF ファイルでは 3 分岐に見えることがあるが,2 分岐になっているはず.
rerooted <- root(MyTree,outGroup,r=T)
rerooted <- ladderize(rerooted)
plot(rerooted,use.edge.length = FALSE)
# rerooted をテキストで保存する.
# Phylip 形式.
#write.tree(rerooted, file="MyTree.trererooted.nwk")
# Nexus 形式
#write.nexus(rerooted, file="MyNexusTreefile.nex")
# dev.off という関数でデバイスを閉じる.
dev.off()
|
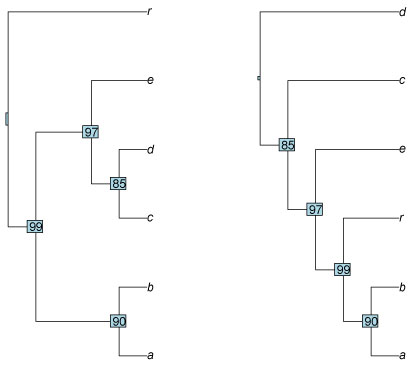
|
| ape: BS 値付き tree の reroot はおかしい? |
reroot すると,BS 値の位置がおかしくなると聞きました.以下は例題です.左の tree を d という leaf で rooting しています.しかし,私には問題ないように思えます.
rerootBS_fol.tar.gz
library(ape)
layout(matrix(1:2, 1, 2))
t=read.tree("test.tre")
plot(t)
nodelabels(t$node.label)
rerooted <- root(t,"d",r=T)
plot(rerooted)
nodelabels(rerooted$node.label)
|
|
| ape: BS 値付き NJ tree を求める |
NJ tree を求めて,ブートストラップ解析を行い,BS 付き NJ tree を返すスクリプトです.BS 解析は 3 つのコドンをまとめて一つとしてサンプリングします.dist.dna は GTR に対応していないため,最もパラメーターリッチである TN93 を用いています.
かつて ape には DNAmodel という関数があって partitioned モデルに対応していたようですが,現在は廃止されています.このため BS 解析以外に partition は行っていません.2011 年に出た phangorn に譲ったようです.
phangorn については,このページの下の方にメモを書きました.
2013 年頃に,boot.phylo のフォーマットが変更になったようです.
njBS.tar.gz
(2024 年 11 月改定)
njsFromSeqFile.tar.gz
|
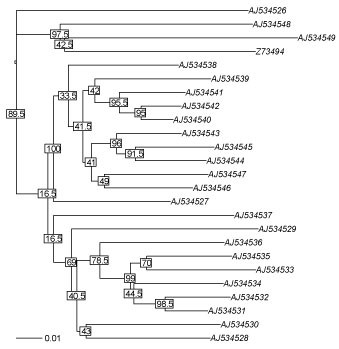
|
コマンドラインバージョン |
 |
115_treePlotB2.zip
2025 年 11 月
|
| ape: 内群根幹の分岐に BS 100% を付ける |
RAxML の BS 付き tree は内群根幹 (外群直下) の分岐に BS 値が付けられていません.Notung の Rearrangement 解析などでは,この分岐を解消してしまうことがあるので,以下のスクリプトで 100% の BS 値を付けると便利です (2018 年 8 月).
rootBS100_fol.tar.gz
$ ls
RAxMLtree.tre rootBS100.R
junINOUEpro@inouejun-no-MacBook-Pro|~/Downloads/rootBS100_fol
$ R
R version 3.3.2 (2016-10-31) -- "Sincere Pumpkin Patch"
Copyright (C) 2016 The R Foundation for Statistical Computing
Platform: x86_64-apple-darwin13.4.0 (64-bit)
.....
> source("rootBS100.R")
|

|
| ape: 配列の違いを距離行列にする |
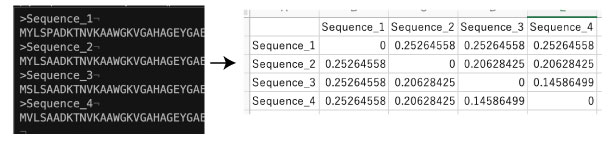 |
library(ape)
library(seqinr)
# アミノ酸配列を読み込み
fasta_file <- "sequences.fasta"
seqs <- read.fasta(fasta_file, seqtype = "AA")
# seqinr の alignment オブジェクトに変換
alignment <- as.alignment(nb = length(seqs),
nam = names(seqs),
seq = sapply(seqs, function(x) paste(x, collapse = "")))
# 距離行列を計算
dist_matrix <- dist.alignment(alignment, "identity")
# "identity" 完全一致の割合に基づく距離(1 - 一致率)
# "similarity" 類似アミノ酸も考慮した距離
# "score" アライメントスコアに基づく距離
# 他の距離法や、モデルベースの方法(例:JTT, WAG)は、phangorn パッケージ。
#### 距離行列を保存
#距離行列を行列形式に変換
dist_mat <- as.matrix(dist_matrix)
# as.matrix() は dist オブジェクトを通常の行列に変換します。
# CSV ファイルとして保存
write.csv(dist_mat, file = "distance_matrix.csv", row.names = TRUE)
#### 系統樹を推定
# 系統樹を作成
tree <- nj(dist_matrix)
# 系統樹を描画
plot(tree, main = "Amino Acid Phylogenetic Tree")
ape_diffCount.zip
2025 年 5 月
|
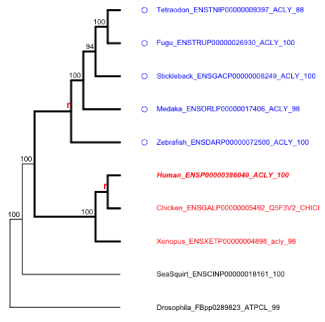
| ape: Tree drawing with color -1 |
 |
treePlotB.tar.gz (2018 年 6 月)
|
| ape: Tree drawing with color -2 |
This script draws a tree into a png file and gives color to specific leaves and labels.
treePlot.tar.gz
Move into the working directory and execute the treePlot.R file.
setwd("/Users/inouejun/Desktop/treePlot")
source("treePlot.R")
(Dec. 2017) |
|
| ape: Tree drawing with thick branches |
|
| ape: Drawing the nhx format tree (Notung output) |
Get into the uncompress nhxReader.tar.gz file and type
perl nhxReader.pl > log.txt
rscript will read a R script, nhxReader.R. To convert nhx to newick format, I am using perl script. Colon within the node label [&&NHX:S=Percomorpha:D=N:B=100.0] should not be used for ape, read.tree function.
Note that the rscript should be the same version of your R. Otherwise, rscript cannot recognize the input file as follows:
[junINOUEpro:newickTreePlot_fol]$ rscript plotNewick.R MyTree.tre
Rscript execution error: No such file or directory
(Dec. 2017)
|
|
sub nhxToNewickNHX {
my ($tree) = @_;
while ($tree =~ /(\[&&NHX:.*?\])/) {
my $exp = $1;
$exp =~ s/&&NHX://g;
$exp =~ s/:/_/g;
$tree =~ s/\[&&NHX:(.*?)]/$exp/
}
#Delete the branch lengths
$tree =~ s/\:[\d|\.E-]+//g;
#Delete the exps with leaves
$tree =~ s/\[S=[a-zA-Z]+\]//g;
return $tree;
}
|
|
|
| phangorn |
インストール
こちらをご覧下さい.5 分ぐらい時間がかかりました.
マニュアル
こちらとこちらです.
|
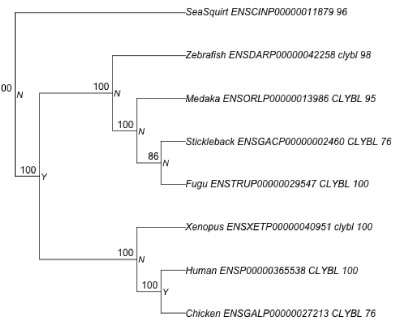
| phangorn: AA 配列から BS 値付き NJ tree を得る. |
ape ではアミノ酸配列から NJ tree などを構築できないようなので,phangorn でやってみました.スクリプトを走らせる前に,「install.packages("phangorn")」によって,パッケージをインストールするのを忘れないでください.ただし,データによってはうまく動かないです.Ape を使って cDNA 配列 (1st, 2nd) で解析した方が良いように思えます (2017 年 1 月).
njBS_Phangorn.tar.gz
|
|
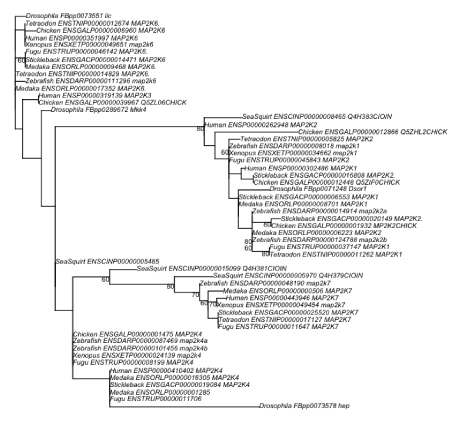
| phangorn: AA 配列から NJ tree を得る. |
アミノ酸配列に基づいて rooted NJ tree を得ることができます.ここではクエリ配列を指定してボールドにしています.
njAAtree_Phangorn.tar.gz
|
|
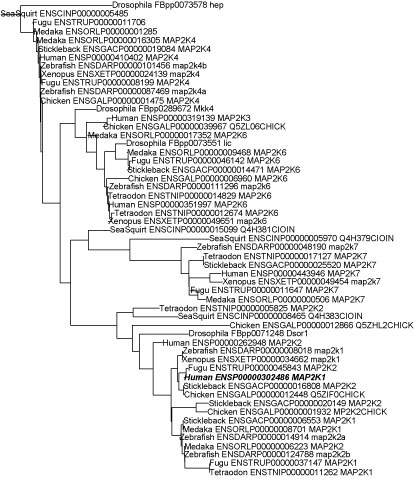
| 年代推定 |
r8s の解析を APE で行います.こちらをご覧ください.ただし,こちらによると chronogram は使われていないため,chronopl を使う必要があるみたいです.しかし以下のステップでは,枝の長さが違う tree を ultrametric tree にするだけで,まだ制約の付け方や時間軸の着いた tree 表示のやり方はわかりません.
##################
library (ape)
# ない場合は,install.packages("ape")
# get tree
data("landplants.newick") # example tree in NH format
landplants.newick
[1] "(Marchantia:0.033817,(Lycopodium:0.040281,......:0.012469):0.019372);"
tree.landplants <- read.tree(text = landplants.newick)
# plot tree
tree.landplants
plot(tree.landplants, label.offset = 0.001)
# estimate chronogram1 [lambda を 1 にしました]
chrono.plants <- chronopl(tree.landplants,1)
# plot
plot(chrono.plants, label.offset = 0.001)
# value of NPRS function for our estimated chronogram [これは動きませんでした]
NPRS.criterion(tree.landplants, chrono.plants)
|
|
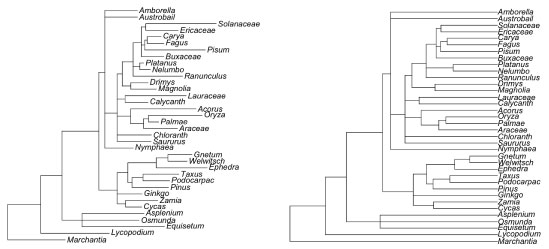
| TreeSim |
TreeSim の getx や corsim を使うことで,全種を網羅できていない系統樹の種分化パターンをシュミレートするソフトウェアのようです.こちらのような解析ができるそうです (T.K さんより).R.3.0.2 以上でないと動かないみたいです.
http://cran.r-project.org/web/packages/TreeSim/TreeSim.pdf
にある例題を試しました.
> numbsim<-3
> age<-10
> lambda<-0.3
> mu<-0
> K<-40
> tree<- sim.bd.age(age,numbsim,lambda,mu,mrca=FALSE,complete=FALSE,K=K)
エラー: 関数 "sim.bd.age" を見つけることができませんでした
> LTT.plot(c(list(tree),list(c(age,age,age))))
エラー: 関数 "LTT.plot" を見つけることができませんでした
上のようになるはずです.以下を実行しました.
> install.packages("ape")
> install.packages("geiger")
> install.packages("TreeSim")
> library("ape")
> library("TreeSim")
するとうまく動きました.
> numbsim<-3
> age<-10
> lambda<-0.3
> mu<-0
> K<-40
> tree<- sim.bd.age(age,numbsim,lambda,mu,mrca=FALSE,complete=FALSE,K=K)
> LTT.plot(c(list(tree),list(c(age,age,age))))
>
PDF ファイルが出ました
|
繰り返し計算と作図の例
library("ape")
library("TreeSim")
iterations <- 10
x <- 3
while (x <= iterations) {
x <- x + 1
numbsim <-3
age <-10
lambda <-0.1 * x
mu<-0
K<-40
tree<- sim.bd.age(age,numbsim,lambda,mu,mrca=FALSE,complete=FALSE,K=K)
par(new=T)
LTT.plot(c(list(tree), list(c(age,age,age)), add=TRUE))
}
|
 |
| 誤差 |
expm1()
以下の二つは数学的に同じ数値を返すはずです.しかし,値が極端に少ないときは,浮動小数点に関するコンピュータの計算能力のエラーからおかしな答えが得られます.こちらによると,expm1(x) の方が exp(x)-1 に比べて正確な値を返すようです.
> expm1(1e-14)
[1] 1e-14
> exp(1e-14)-1
[1] 9.992007e-15
大きな値で計算すると,同じ答えが得られます.
> expm1(0.1)
[1] 0.1051709
> exp(0.1)-1
[1] 0.1051709
|
| Bioconductor のインストール |
R prompt で以下のコマンドを入力してください.
source("http://bioconductor.org/biocLite.R")
有用なモジュールの追加.
biocLite("edgeR")
biocLite("DESeq")
biocLite("etc")
biocLite("Biobase")
install.packages("gplots")
install.packages("ape")
|
| hypot() |
Hypotenuse (斜辺) Function
こちらをご覧下さい (2014 年 11 月).
|
| expm1() |
こちらをご覧下さい (2014 年 11 月).
exp(x) = 1 + x + x^2/2 + x^3/6 + ....
exp(x) -1 = x + x^2/2 + x^3/6 + ....
|
| pheatmap |
| Error: unable to open connection to X11 display |
R の cairo 問題と名付けました。
Ubuntu24.04。R, 4.4.1 で遭遇。R のコンソールで png() と入力するとこのメッセージが現れました (2024 年 7 月)。
In png(png.file, width = pngWidth, height = pngHeight) :
unable to open connection to X11 display
以下を R のコンソールに入力すると、cairo というライブラリが FALSE になっています。
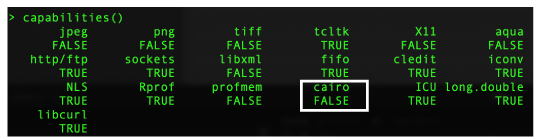
library() によってライブラリ一覧すると cairo がインストールされていることがわかるので、この問題は、cairo が入っていても R が認識していないために生じているようです。
解決方法1: pdftoppm で pdf から png を生成する
どうしても解決しないので、pdftoppm で対応しました (2024 年 8 月)。
pdftoppm -png -scale-to 1024 115_1stGeneTree.pdf > 115_1stGeneTree2.png
|
解決方法2: options(bitmapType='cairo') を記述する
R kernel constantly crashes in Jupyter Notebooks due to the graphics
(2024 年 )
質問者は R 4.3.2 を Ubuntu 22.04 remote machine で走らせている。Jupyter Notebook。エラーメッセージ:
unable to start device PNG
Calls: <Anonymous> ... evaluate -> dev.new -> do.call -> <Anonymous> -> ok_device
In addition: Warning message:
In ok_device(filename, ...) : unable to open connection to X11 display ''
Execution halted
~/.Rprofile に
options(bitmapType='cairo')
を記述するが解決せず、新たに以下のメッセージを得る。
13:52:48.798 [warn] StdErr from Kernel Process Error in dev.control(displaylist = "enable") :
dev.control() called without an open graphics device
Calls: <Anonymous> ... tryCatch -> tryCatchList -> evaluate -> dev.control
In addition: Warning message:
In ok_device(filename, ...) :
13:52:48.798 [warn] StdErr from Kernel Process type = "cairo" is unavailable. trying "Xlib"
Execution halted
png() でも以下のメッセージ
> png()
Error in .External2(C_X11, paste0("png::", filename), g$width, g$height, :
unable to start device PNG
In addition: Warning message:
In png() : could not open PNG file 'Rplot001.png'
そこで、R-4.3.2 をインストール
./configure --with-x=yes --with-libpng=yes --enable-R-shlib
make
sudo make install
その後、~/.Rprofileに以下を記述
options(bitmapType='cairo')
なお、R コンソールで以下を入力すると、cairo や X11 が可能かどうか調べられる。
capabilities()
私はこれで、ようやく R で png() が問題なく動くようになりました。再インストールの際には、このページの上の方に書いておきましたが、アンインストールを行いました (2024 年 7 月)。
=> Ubuntu と R を全く同じバージョンにして他のサーバーで試したのですが、cairo 問題は解決しませんでした。そこで、pdftoppmを使って、pdf から png ファイルを生成することにしました (2024 年 8 月)。
|
| ### 以下、その他参考にしたサイトです。 |
Q&A (初級者コース)/13
(2006 年 11 月)
質問者は、cgi スクリプトから R を起動しグラフを作成する際に、jpeg デバイスを開くときにエラーに遭遇
Error in X11(paste("jpeg::", quality, ":", filename, sep = ""), width, :
unable to start device JPEG
In addition: Warning message:
unable to open connection to X11 display ''
Execution halted
以下、引用:RのJPEG,PNGはX11の動作している環境が必要です. CGIに環境変数DISPLAY=:0.0 をあたえる等してください. この手の問題はUnix系のFAQでXvfbとかでググれば沢山出てくると思います。 |
How to run R scripts on servers without X11 [duplicate]
type = "cairo" is unavailable. trying "Xlib"
への対処。この質問者は、R scripts で プロットを png() で保存したいが、上記のエラーが出た、と質問。X11 は Unnix servers でサポートされていないので
"if you are using R 3.0, try options(bitmapType='cairo') it worked for me"
という答えの記述に、options(bitmapType='cairo') を png の前に書くべし、とある。
|
R Ciro installation without apt-get, sudo
cairo
ソースコードから。git でダウンロード。
|
| nMDS |
| nMDScolors.tar.gz |
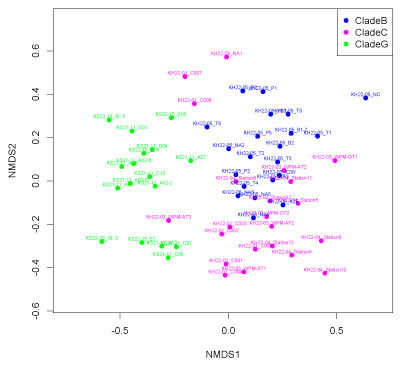 |
| 2025 年 2 月 |
| |
| hclust |
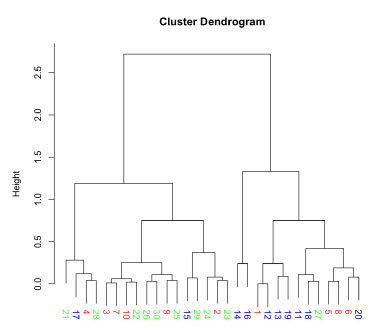 |
# データの準備
h <- hclust(dist(PlantGrowth$weight))
# 系統樹のプロット(ラベルとタイトルを非表示にする)
plot(h, labels = FALSE, xlab = "")
# ラベルの取得
labels <- rownames(PlantGrowth)[h$order]
# グループごとに色を設定
group_colors <- c("red", "blue", "green")
group <- as.factor(PlantGrowth$group)
label_colors <- group_colors[group[h$order]]
# リーフの y 座標を取得
leaf_y <- rep(-0.3, length(labels))
# ラベルを色分けして再描画(リーフの位置に配置)
text(x = 1:length(labels), y = leaf_y, labels = labels, col = label_colors, srt = 270, adj = 0, xpd = TRUE)
ただ、
jc.vegdist <- vegdist(data_binary, method="jaccard", binary=TRUE)
h <- hclust(jc.vegdist)
だと、leaf が出力されないです。
# 2025 年 1 月
|
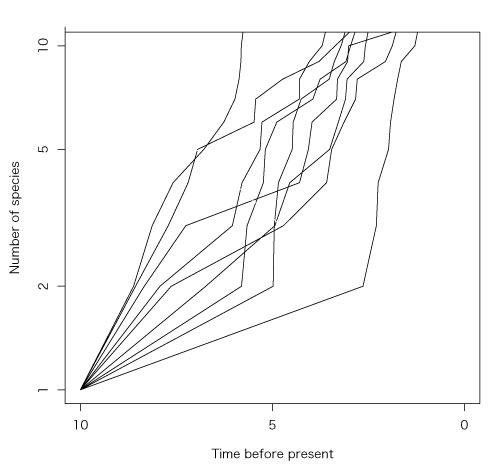
| rarefaction |
iNEXT version
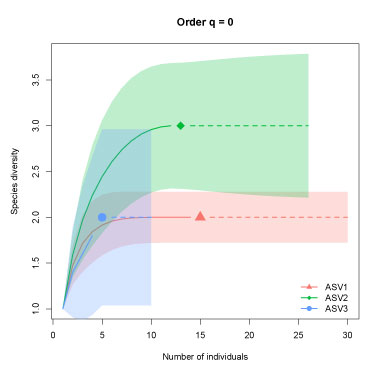
|
| rarefaction_ji.tar.gz |
|
vegan version

|
| rafefuction_vegan_ji.tar.gz |
2025 年 4 月
|
| 指標種 |
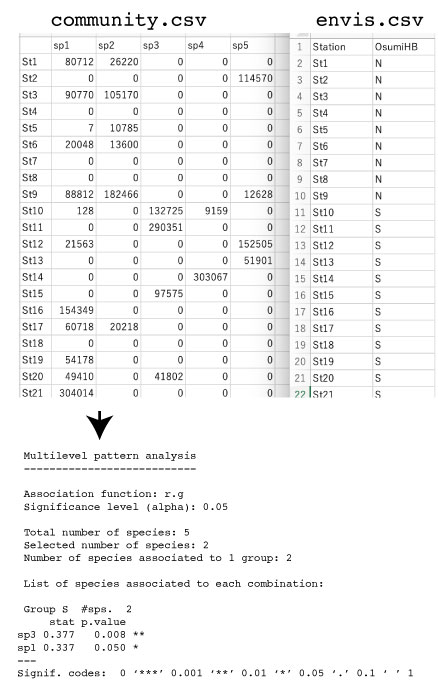 |
| multipatt.zip |
| 2025 年 7 月 |
| |
| HTMLと R で互換性のある代表的な色名 |
| "black" |
|
|
"yellow" |
|
|
"gray" |
|
|
"navy" |
|
|
| "white" |
|
|
"orange" |
|
|
"brown" |
|
|
"lime" |
|
|
| "red" |
|
|
"purple" |
|
|
"pink" |
|
|
"teal" |
|
|
| "green" |
|
|
"magenta" |
|
|
"gold" |
|
|
"olive" |
|
|
| "blue" |
|
|
"cyan" |
|
|
"silver" |
|
|
"maroon" |
|
|
|
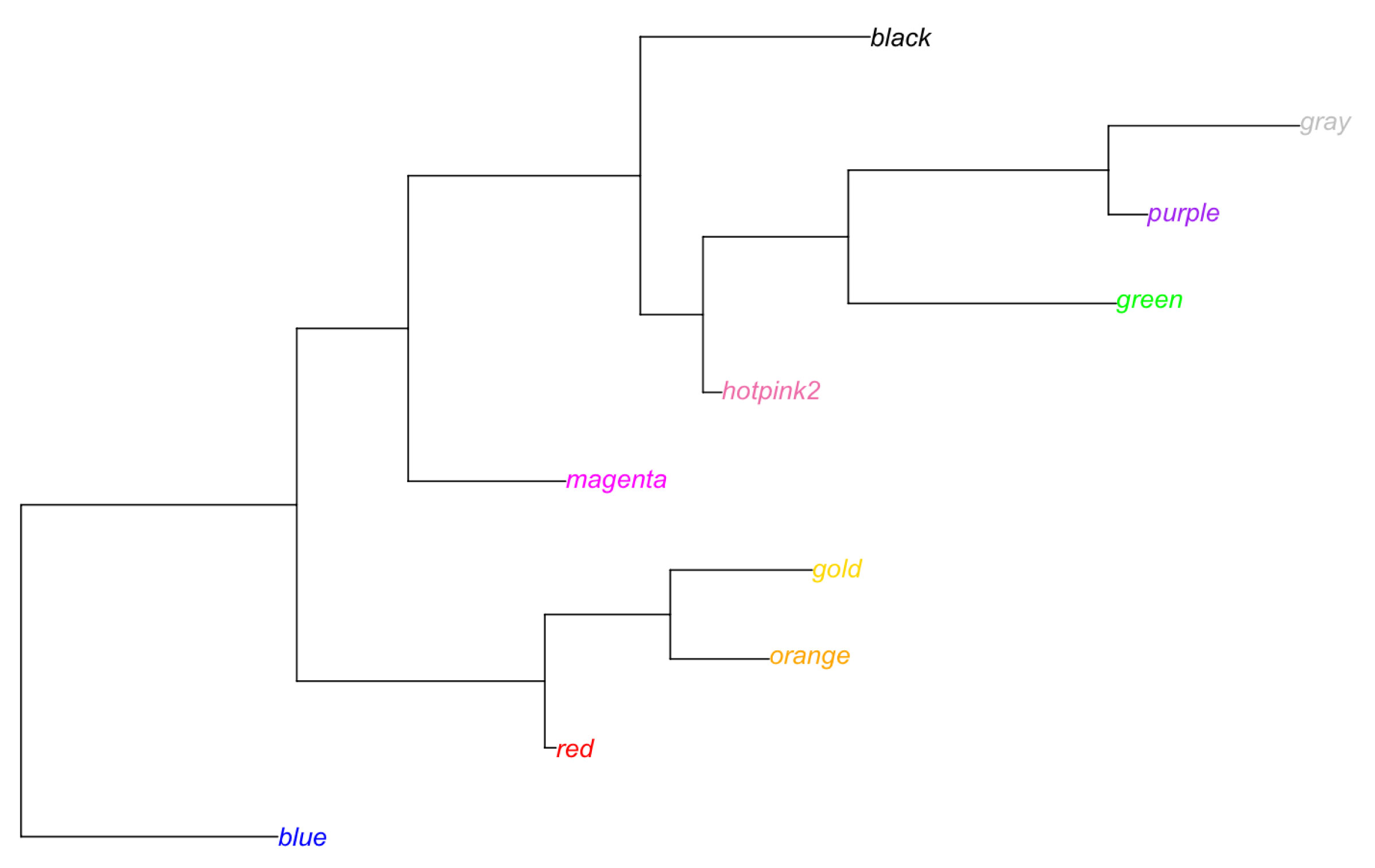
library(ape)
# 色名をラベルとして使う
labels <- c("blue", "red", "orange", "gold", "magenta", "hotpink2", "green", "purple", "gray", "black")
# 系統樹の作成
tree <- rtree(length(labels))
tree$tip.label <- labels
# tip.color にラベルと同じ色名を使う
plot.phylo(tree, tip.color = labels) |
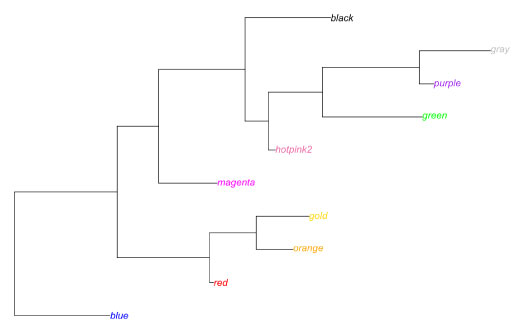 |
| 2025 年 9 月 |
| |
| リンク |
|
|