| 解析パイプライン |
| 基本構造 |
基本的な構造:すべてのタンパク質配列について以下の処理を行います.
1. 候補配列の収集
2. 遺伝子系統樹の推定
3. オーソログの選定

スクリプトをつなげる:以下のように,Perl script の system command を使って,短いスクリプトをつなげます.
for (....) {
system("perl 010SCRIPT.pl infile > 010out.txt");
system("perl 020SCRIPT.pl 010out.txt > 020out.txt");
system("perl 030SCRIPT.pl 020out.txt > 030out.txt");}
| 1. 候補配列の収集 |
二種類のデータ:Ensembl を使います.種ごとにすべての遺伝子について cDNA 配列とアミノ酸配列が公開されています.一つの遺伝子について,cDNA 配列とアミノ酸配列,2 種類の fasta ファイルをダウンロードします.

release-86 > fasta > tetraodon_nigroviridis に入って,以下を参照に cds と pep ファイルをダウンロードしてみましょう.


1.2. トランスクリプトを選ぶ
遺伝子によっては,複数のトランスクリプトが報告されています.スクリプトを書いて,とりあえず最長のトランスクプトを選びます.
1.3. 類似性検索
Local blast を使います. 全アミノ酸配列データを種ごとにデータベース化して,ヒトのアミノ酸配列をクエリに blast 検索します.
| 2. 遺伝子系統樹の推定 |
2.1. アライメント
アミノ酸配列のアライメントは MAFFT を使って,行います.cDNA 配列のアライメントは,PAL2NAL を使います.
2.2. トリミング
アライメント困難な領域の削除は,trimAl を使います.
2.3. 遺伝子系統樹の推定
RAxML を使って最尤樹を推定します.時間がかかるので,Supercomputer を使って並列に解析を行います.近隣結合法は R の Ape パッケージを使います.それぞれブートストラップ解析も行います.
多くの場合,アミノ酸配列より cDNA 配列で解析を行った方が良い系統樹が得られます.アミノ酸より cDNA 配列の方が長い,というのが主な理由だと思います.
| 3. オーソログの選定 |
こちら (sample_recon.tar.gz) の例題を用いて説明します.
3.1. 遺伝子系統樹と種の系統樹を比較
Notung を使って遺伝子系統樹と種の系統樹を比較し,オーソログ・グループ (オーソログの集合) を判定します.
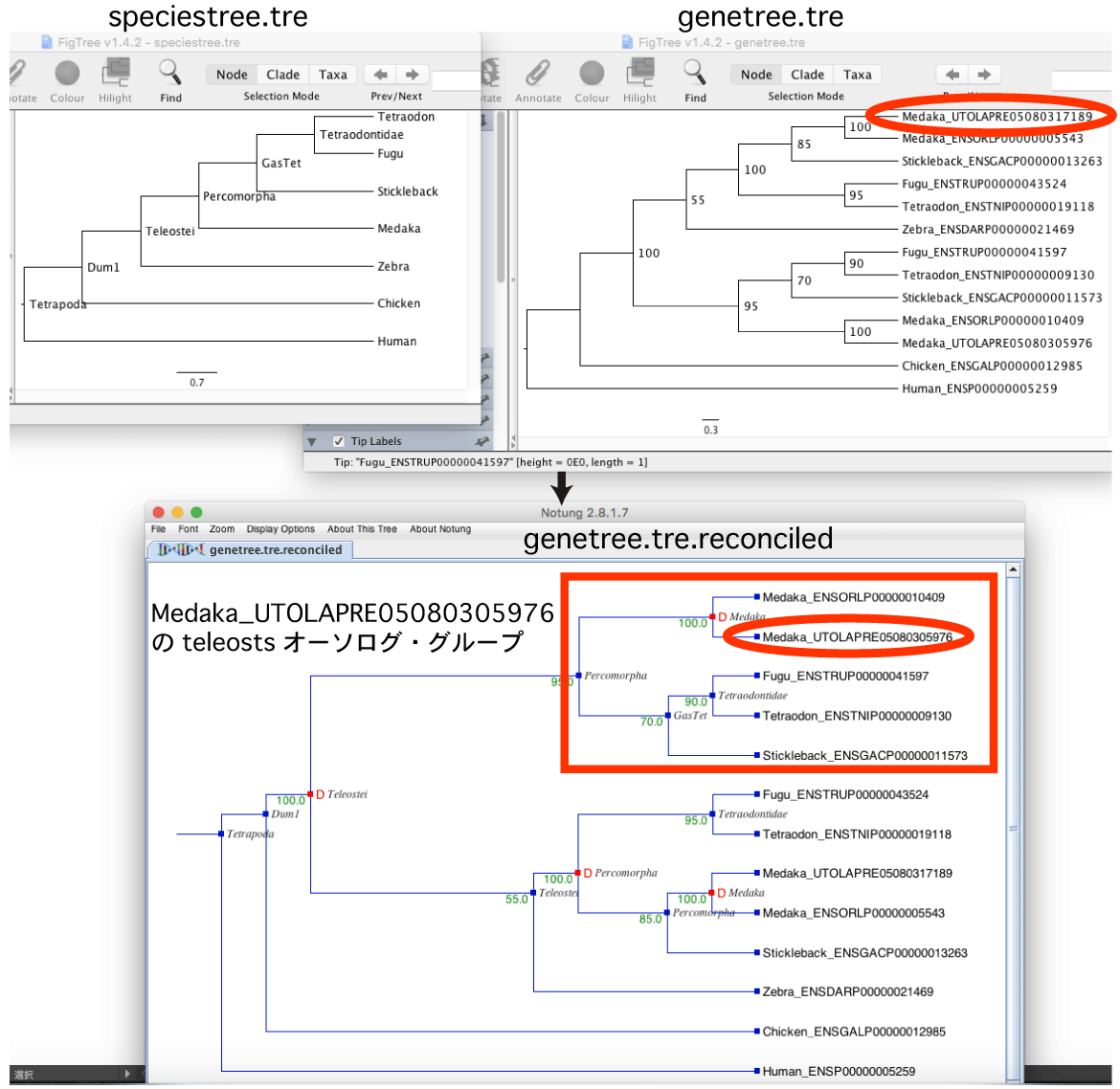
3.2. オーソログ・グループの選定
Perl script で Notung から出力される NHX フォーマット の tree を処理し,以下の条件を満たすクレードを探します.
このとき遺伝子系統樹で推定された枝のブートストラップ確率 (B=95.0) を参考に,信頼性の高い遺伝子系統樹だけを選ぶことも可能です.・クエリ配列 (図では Medaka_UTOLAPRE05080305976) が含まれる.
・Teleostei か Percomorpha というクレードで,もっとも leaf が少ない.
・根幹の分岐が D=N とタグされている.D=Y は遺伝子重複による分岐を示す.
([&&NHX:S=Percomorpha:Nset=<Medaka@GasTet>:H=N:D=N:B=95.0] とラベルされている)
| 3.3. Newick format の描画 R を使います. treePlot.tar.gz を参照してください. |
 |
| 染色体上へのマッピング |
| 得られたオーソログ情報を Circos を使ってマッピングすると,右図のように二種間でゲノム構造の比較ができます.遺伝子の染色体上の位置を得るには,Ensembl の BioMart を使います. |  |



| 右にある 5 つの protein ID (Human) を下図で示した箱にコピー&ペースしてください. | ENSP00000346733 ENSP00000382114 ENSP00000346595 ENSP00000370718 ENSP00000372154 |




|
|