|
|||
|
2024 年 4 月 7 日 井上 潤
|
|||
|
|||
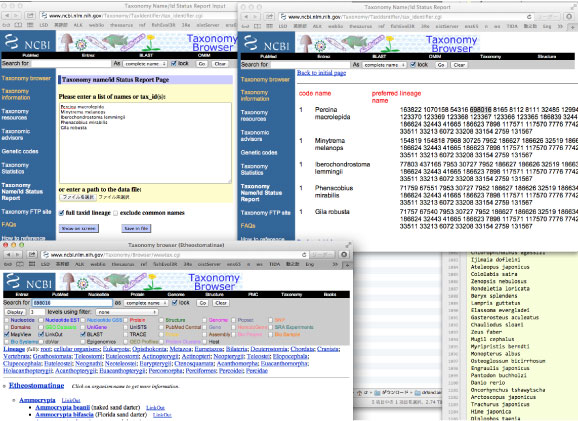
NCBI の Taxonomy Browser を使います.
|
|||
 |
|||
|
|||
| ミトコンドリアゲノムであれば,ふつうは簡単です. 例えば脊椎動物なら,こちらのページから Accession number を押して GenBank format のページまでたどって,1 種づつ GenBank format をあつめても良いでしょう.あとはこのページの下方で紹介している GenBankStrip.pl で GenBank format を処理して遺伝子データセットを得れば良いです.Accession number を一つのファイルに保存しておけば,以下の「学名をリストアップして一気にデータを集める」に書いている Batch Entrez を使った方法で GenBank format になってミトコンドリアゲノムをたくさんの種から一気に集めることができます. |
|||
| 以上の方法で集められない場合は,これから紹介する方法が考えられます. Entrez を使って,必要な配列を検索して一気にダウンロードすることもできます.まずは TOGO TV を見た方が良いです. 以下にいくつかの検索例を示します.例題で用いたコマンドの説明は,Entrez Nucleotide, Help に行くと Entrez Help (PDF ファイル) として配信されています.PDF であれば,12 ページの "Range Searching" あたりを見ると,だいたい把握できると思います.それ以外は Acrobat Reader の find を使って,"PDAT" などを検索して拾い読みして下さい.便利そうな例をマニュアルで見つけたので,以下のコラムに張っておきます. |
|||
Example searches: |
|||
| 700 個体以上のヒト・ミトコンドリアゲノムデータを集める | |||
|
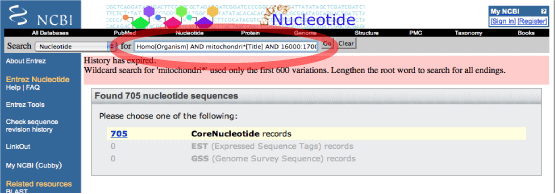
たとえば,ヒトのミトコンドリア全長配列だけを集めたい場合は,以下のコマンドを赤い丸で囲ったウィンドウに入力して下さい.ポップアップメニューは "Nucleotide" にします.リンクの張られた数字 (705) をクリックすると,リストが表示されます.クエリフィールドについてはこちらをご覧下さい.
|
|||
 |
|||
|
リストアップされた全てのファイルをダウンロードするには,ポップアップメニューを "GeneBank" にして画面が変わるのを待ち,"Send to" となっているポップアップメニューを File にして下さい."sequences.gb" というファイルがダウンロードされます.GeneBank フォーマットから遺伝子を切り出すには,GenBankStrip.pl を使うと良いでしょう.詳しくはすぐ下のコラムを参照して下さい. |
|||
| ある期間に登録されたミトコンドリアゲノムデータを集める | |||
|
|
|||
| 条鰭類のミトコンドリア遺伝子配列データを集める |
|||
|
"NOT Nuclear[Title]" を入れないと,核にコードされていると思われる余分な配列を集めてしまうようです.
GenBankStrip.pl で処理すると,以下のような結果が得られました.ここで注意したいのが,"500:20000[SLENG] です.500~20000 bp の部分配列を含めた配列を示しますが,~10000bp と設定してしまったりすると,ミトコンドリアゲノム配列として登録されている配列が除外されてしまいます.
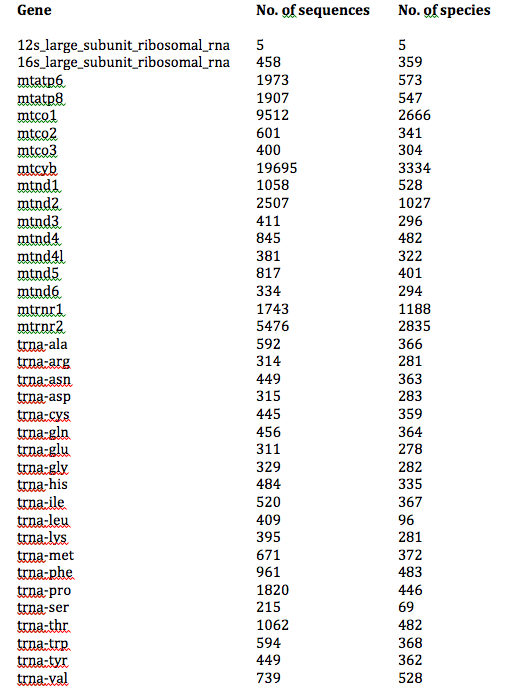
数レコードしか得られなかった遺伝子は除いてあります (たくさんありました).また,12S が 5 個しか得られないなど,後で改善すべき問題点もあります. |
|||
|
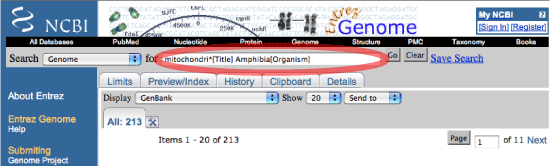
ミトコンドリアゲノム配列だけを集める場合は,Search のポップアップメニューを Genome にすれば,より精度良く分類群を指定してデータを集められます.ただし,Genome にするとヒトのように同種の個体別データを集められなくなります.通常のミトコンドリアゲノム解析であれば,種ごとにデータを集めることになるので,Genome にしてデータ収集を行った方が楽です.ここでも期間限定などの制約をかけてデータを集めることができます."mitochondri*[TItle]" を入れないと,核ゲノムのデータも出てくるので注意して下さい.
目的とする数のデータが集められたかどうかは,Organelle Genome Resources のページで確認すると良いです. |
|||
 |
|||
| 学名をリストアップして一気にデータを集める | |||
|
Batch Entrez に行って手順通りやれば簡単にできます. |
|||
Polyodon_spathula |
|||
| Accession number をリストアップして一気にデータを集める | |||
| 以上と同様の手順です. | |||
|
|||
| その他 | |||
|
|||
|
|||
|
Perl script.複数種の GeneBank フォーマットから,遺伝子別に配列を切り出します.それほど詳細ではないですが,使用方法は script の最初に書いてあります.
Perl (v5.34.1) では動かなかったです。以下のエラーメッセージがでます。 "Can't use 'defined(@array)' (Maybe you should just omit the defined()?) at GenBankStrip.pl line 280." Perl の仕様が変化したためらしいです。 以下、GenBankKiridashi.tar.gz で代用できるかお試しください (2024 年 4 月)。 1) NCBI の webpage にある Search (Nucleotide などに設定) を使って,"Actinopterygii RAG1" あるいは,"Actinopterygii mitochondrion"のように遺伝子配列をキーワード検索します. 2) GenBank フォーマットで検索結果をダウンロードします.NCBI のサイトの pull down メニューを操作することで,まとめたファイルを自動的にダウンロードできるので,手動でテキストファイルに copy & paste する必要はありません. 3)
切り分けた結果は,seq_gbs.striplist.txt に書かれているので,まずはこれを見てねらい通りのデータセットが作成されたかを確認します. |
|||
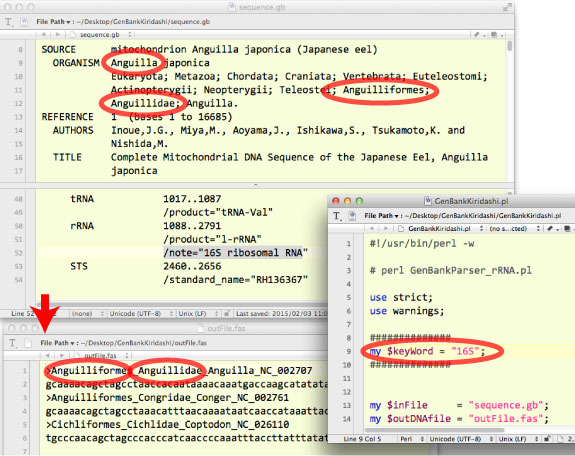
| GenBankStrip.pl の条鰭類 mtGenome 限定バージョンです.分類群情報が、アウトプットとして得られる fasta の name line に入ります (2015 年 2 月 井上). | |||
 |
|||
| 例題:GenBank フォーマットから,RAxML インファイルをつくる | |||
| こちらに書きました (2018 年 8 月). | |||
|
|
|||
|
Olaf R.P. Bininda-Emonds さんが作成した perl script.Nexus 形式で保存したシーケンスファイルを fasta 形式にしてくれます.MacClade を用いて遺伝子配列を concatenate するには,fasta から NBRF 形式にする必要があります (最近は fast でも concatenete できるようになりました).MacClade でもできますが,unix (ターミナル) を用いた方が簡単にできます.
基本的な操作 seqConverter と Nexus 形式の infile を同じフォルダに入れておきます.terminal でこのフォルダに移動し,
と入力します.infile.phylip というファイルが作成されるので,これを BBEdit などのエディターを用いて改行コードを変換します.
を加えます.
|
|||
|
|
|||
|
BioPerl を使って,GenBank からミトコンドリア遺伝子の配列を自動的に切り出してダウンロードしてくれるソフトウェア.私は Perl 初心者なのでまだうまく動かせないでいますが,ミトコンドリア遺伝子を扱う研究者にとっては便利なアプリケーションなのではと期待しています.以下の論文で登場を知りました.
|
|||
|
|
|||